This guide contains general technical information for working with animal cells in culture, including media, subculturing, cryopreservation, and contamination.
Staying Safe in a Pandemic Environment
Getting Started with an ATCC Cell Line
Cell Growth and Propagation
Complete Growth Media
Culture Vessels and Surfaces
Cryopreservation
Contamination and Biosafety
ATCC Media, Sera, and Reagents
Glossary
References
Download a PDF of our Animal Cell Culture Guide
Download NowStaying safe in a pandemic environment
When the recent coronavirus pandemic hit, laboratories throughout the world resolved to shut down operations, reduce the scale of work, or proceed at full steam. To safeguard the health of our scientists, ATCC has adopted a battery of best practices that minimize transmission of SARS-CoV-2 with little impact on productivity. Whether returning after a hiatus or gearing up for a new project, we can all use a refresher to help follow best practices. Please read this first section of the culture guide for some quick reminders about common contamination hotspots and advice on how to keep them in check while getting your work done.
Materials, contamination, and workspace
You may be just getting back into the laboratory or beginning a new project. Before you start, consider some potential hotspots that can profoundly affect your experimental results, such as the quality of your starting materials, execution of proper laboratory technique, and organization of your workspace. Here are some simple tips and techniques to avoid ruining your experiments, leading to confounding results, paper retractions, financial loss, and damaged reputation.
Material for research
- Check existing materials for signs of contamination.
- Authenticate and replenish your cell lines and microbes.
- Start new projects with trustworthy materials.
- Routinely check the expiration dates of media and reagents.
Cross contamination
- Ensure everyone—new and experienced—is trained on aseptic techniques.
- Aliquot your samples and reagents.
- When aliquoting is impractical, put just the amount of the reagent you expect to use into a secondary container. Discard the remainder when finished working.
- Avoid sharing pipettes or other equipment.
- Clean the insides and exteriors of pipettes and tools that must be shared.
- Utilize the biosafety cabinet to reduce contamination.
Personal space and equipment
- Keep bench space uncluttered.
- Wash your lab coat regularly.
- Leave personal items outside.
- Clean your work area before and after use.
- Use lab tablets instead of personal phones.
- If personal items are needed, sanitize them before and after lab use.
- Move extra equipment away from walls and crevices to facilitate frequent and thorough cleaning.
Avoiding infection
Like you, we’re committed to protecting the health of our colleagues. While SARS-CoV-2 is currently unique in its pathogenic nature and transmission dynamics, other infectious organisms may in time arise to threaten the health of laboratory workers. To reduce the chance of contracting a current or emerging infectious disease while working in the lab under epidemic or pandemic conditions, we recommend you follow these best practices.
Coming and going
- Wash your hands well when entering and leaving the lab.
- Master the basics of proper personal protective equipment (PPE) use and removal.
- Stay home if you’ve been exposed to any illness.
- Inspect PPE prior to use.
Interacting with others
- Remember, particles spread via talking, coughing, and breathing.
- Use virtual collaboration tools, and only converse before or after working on cell cultures.
- Always keep your nose, mouth, and skin covered with PPE.
- Be extra vigilant about PPE use when working with animals.
- Keep 6 feet of space between individuals.
Airborne transmission
- Reduce foot traffic in the lab.
- Designate one-way traffic flows to support distancing.
- Try limiting capacity to aid physical distancing.

Getting started with an ATCC cell line
ATCC cell lines and hybridomas are shipped frozen on dry ice in cryopreservation vials or as growing cultures in flasks at ambient temperature. Upon receipt of frozen cells, it is important to immediately revive them by thawing and removing the DMSO and placing them into culture. If this is not possible, store the cells in liquid nitrogen vapor (below −130°C). Do not store frozen cells at temperatures above −130°C as their viability will decline rapidly.
Product sheet
ATCC cell line Product Sheets that contains detailed information for handling the cells may be found at the ATCC website or contact ATCC Product Experience to request a copy. The Product Sheet also contains batch-specific information such as the number of cells per vial, the recommended split or subcultivation ratio, and the passage number when known.
Preparation of medium
Prepare for reviving cell lines by assembling the appropriate medium, serum, and additional reagents required for growth. Many of these products are available from ATCC and can be ordered with the cell lines. These are the same reagents used by ATCC for cell growth and preservation. (See: NOTE 1)
NOTE 1
While most cell lines can replicate in more than one culture medium, their characteristics may alter when the medium is changed. For this reason, starting cell cultures in the same medium used by ATCC is recommended for the best results (see the Product Information Sheet and ATCC website). For details on adapting a cell line to a new medium, see Adapting to a new medium or serum.
Initiating frozen cultures
- Prepare a culture vessel so that it contains the recommended volume of the appropriate culture medium as listed on the Product Sheet, equilibrated for temperature and pH (CO2).
- Thaw the vial by gentle agitation in a water bath at 37°C or the normal growth temperature for that cell line. Thawing should be rapid, approximately 2 minutes or until ice crystals have melted.
- Remove the vial from the water bath and decontaminate it by dipping in or spraying with 70% ethanol. Follow strict aseptic conditions in a laminar flow tissue culture hood for all further manipulations.
- Unscrew the top of the vial and transfer the contents to a sterile centrifuge tube containing 9 mL of the recommended medium. Remove the cryoprotectant agent (DMSO) by gentle centrifugation (10 minutes at 125 × g). Discard the supernatant, and resuspend the cells in 1 or 2 mL of complete growth medium. Transfer the cell suspension into the culture vessel containing the complete growth medium and mix thoroughly by gentle rocking.
- Examine the cell cultures after 24 hours and subculture as needed. (See: NOTE 2)
NOTE 2
Some cell lines, such as hybridomas, take several days before fully recovering from cryopreservation. Some hybridomas have poor viability the first day in culture and will generate cellular debris. After this point, the cells will begin to recover and enter exponential growth.
Processing flask cultures
Some ATCC cell, are shipped as growing cultures in culture vessels. These vessels are seeded with cells, incubated to ensure cell growth and then filled completely with medium for shipping.
Upon receiving a flask culture, visually examine the medium for macroscopic evidence of microbial contamination. This includes unusual pH shifts (yellow or purple color from the phenol red), turbidity, or particles. With an inverted microscope at low power (100×) check the medium for evidence of microbial contamination as well as the morphology of the cells. See more details on examining cell cultures.
If the cells are attached and growing in a monolayer:
- Aseptically remove all but 5 mL to 10 mL of the shipping medium. The shipping medium can be saved for reuse and should be stored at 4°C.
- Incubate the flask at the temperature and CO2 concentration recommended on the Product Sheet (37°C with 5% CO2 for most cell lines) until the cells are subcultured.
If the cells are not attached or are growing in suspension:
- Aseptically transfer the entire contents of the flask to a centrifuge tube.
- Centrifuge at 125 × g for 5 to 10 minutes.
- Remove all but 10 mL of the shipping medium supernatant and resuspend the cells. Store the remainder of this medium at 4°C for later use.
- Aseptically transfer the resuspended cells to a 25-cm2 flask or 75-cm2 flask, depending upon the cell line (see the Product Sheet).
- Incubate the cells at the temperature and CO2 concentration recommended on the Product Information Sheet until cells are subcultured.
Most cell lines begin as primary cultures originating from a piece of minced or enzyme-dispersed tissue. Primary cultures, as mixtures of several cell types, retain the characteristics of their source tissue.
After a period of time, primary cultures will reach confluency, the state when all available space of the culture vessel is covered due to cellular expansion. At this point, the culture will need to be disaggregated (usually with proteolytic enzymes like trypsin) into individual cells and subcultured (split, passaged, or transferred). Following this first passage, the culture is generally referred to as a cell line. With each subsequent subculture, the cellular population becomes more homogeneous as the faster growing cells predominate. Cells with desired properties can also be selected out of the culture by cloning.
Diploid cell lines rarely progress beyond a few population doublings. They have a finite replicative capacity and begin to slow down and eventually stop dividing after 20 to 80 population doublings.1 Recent evidence suggests that some of the observed cellular senescence in cell culture may be due to inappropriate culture conditions as opposed to a predetermined replicative senescence.2 Still other data support replicative senescence for the cells of some species (notably human) even when grown in improved culture conditions. This senescence is mediated by the shortening of the ends of the chromosomes (telomeres) with each cell division.3
In contrast, continuous (or immortalized) cell lines have infinite replicative capacity. These lines are derived from cell lines through immortalization or transformation by any one of a number of means. Many continuous cell lines were derived from tumor tissue. Most of the cell lines in the ATCC collection are continuous, though a few, such as CCD-1117Sk human skin fibroblast (ATCC CRL-2465) or CCD-18Co human colon (ATCC CRL-1459) are finite.
As noted in the section on culture vessels, cell lines grow either attached to a surface (anchorage dependent) or in suspension (anchorage independent). As cells grow and divide in a monolayer or in suspension, they usually follow a characteristic growth pattern composed of four phases: Lag, log or exponential, stationary or plateau and decline.
- Lag phase — Immediately after seeding of the culture vessel, the cells grow slowly while recovering from the stress of subculturing.
- Log or exponential phase — The cells enter a period of exponential growth that lasts until the entire growth surface is occupied or the cell concentration exceeds the capacity of the medium.
- Stationary phase — Cell proliferation slows and stops.
- Decline phase — If the culture medium is not replaced and the cell number is not reduced, the cells lose viability and their number decreases.
To ensure viability, genetic stability, and phenotypic stability, cell lines need to be maintained in the exponential phase. This means that they need to be subcultured on a regular basis before they enter the stationary growth phase, before a monolayer becomes 100% confluent or before a suspension reaches its maximum recommended cell density. Generating a growth curve for each cell line is useful to determine the growth characteristics of the cell line. (See: Figure 1)
For detailed information on the growth and propagation of any ATCC cell line, see the specific cell line Product Sheet which can be found on the ATCC website, or contact ATCC Product Experience to have one sent to you.

Cell growth and propagation
Passage number and population doubling level
Primary cultures are generally subcultured at a 1:2 ratio (they are split in half with each passage). Most continuous cell lines replicate at higher rates and are subcultured at a much higher split ratio. Passage number is generally the number of times the cells have been subcultured into a new vessel. For diploid cultures, passage number is roughly equal to the number of population doublings (or population doubling level, PDL) since the culture was started. This is not the case for continuous cell lines as they are passaged at higher split ratios. Consequently the PDL is not determined for continuous cell lines. In most cases, the PDL is an estimate as it does not account for any cells that were lost due to death from necrosis or apoptosis or cells which are nearing senescence and no longer divide. Calculate the population doubling level with the following formula:
PDL = 3.32 (log Xe – log Xb) + S
Xb is the cell number at the beginning of the incubation time.
Xe is the cell number at the end of the incubation time.
S is the starting PDL.
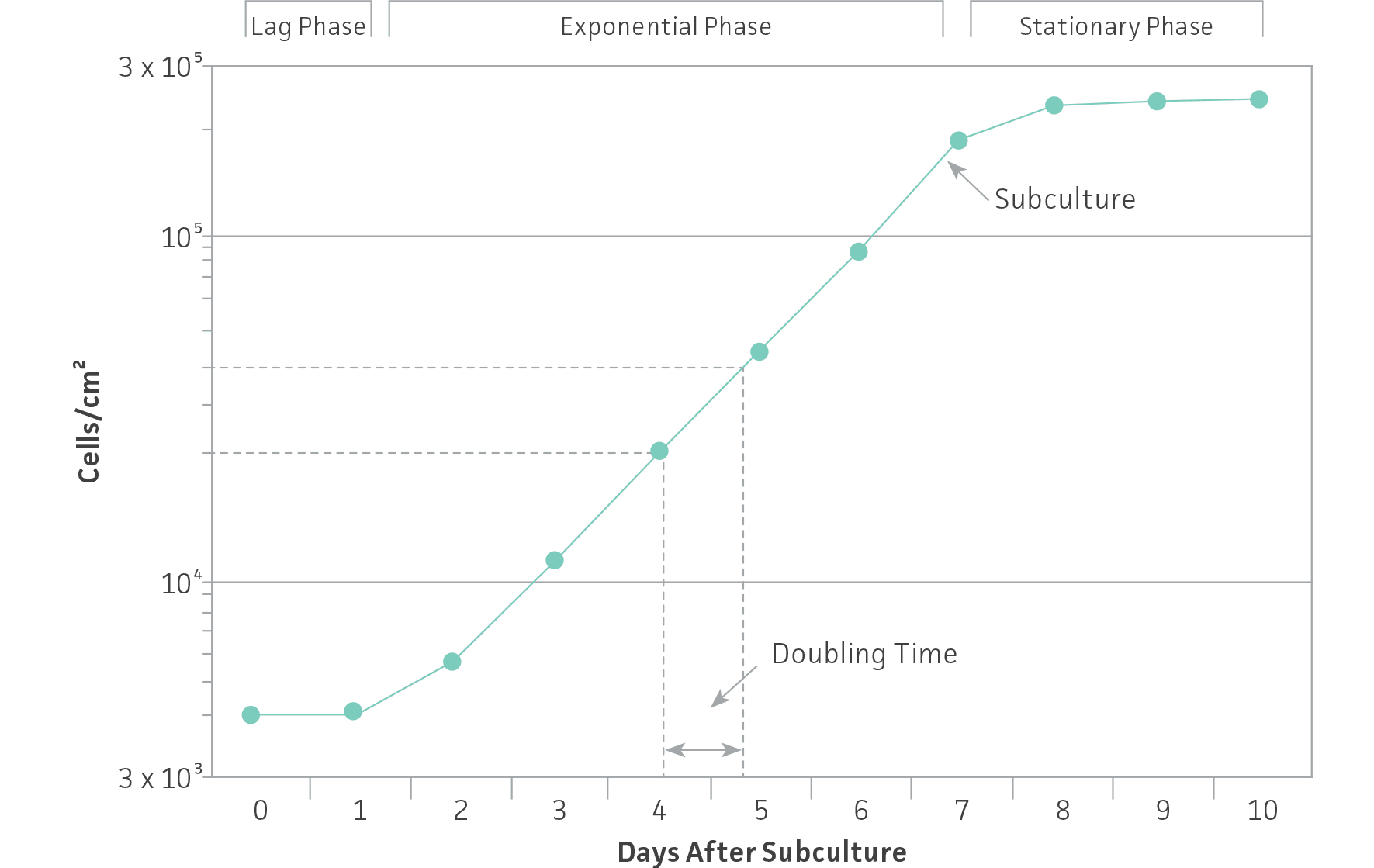
Figure 1: Growth curve for cells grown in culture. Cells should be subcultured while still in the exponential phase.
Calculate the population doubling time, or the time required for a culture to double in number, with the following formula:
DT=T ln2/ln(Xe/Xb)
T is the incubation time in any units.
Xb is the cell number at the beginning of the incubation time.
Xe is the cell number at the end of the incubation time. (See: NOTE 3)
ATCC tracks the PDL and passage number for many adherent cell lines when the depositor supplies this information at the time of deposit. See the Product Information Sheet for the specific cell line for the passage number and/or PDL as part of the batch-specific information supplied.
NOTE 3
Cells grow at different rates in each of the different phases of the growth cycle and the calculated doubling time may be a composite of growth during more than one of these phases. Growth during exponential growth or log phase is fairly constant and reproducible for a given set of growth conditions.
Adapting to a new medium or serum
To ensure that the characteristics of your cell line remain constant, maintain your cells in the same medium, serum, and supplements with the same subculturing regimen used to establish the culture. Any change to the culturing conditions has the potential to change the characteristics of the cell line.
Be particularly cautious when working with a new cell line as media formulations vary among suppliers, even for media with similar or identical names. Read descriptions, formulations, and labels carefully to ensure that the appropriate medium is used or the cell line may be inadvertently adapted to a new medium. All ATCC cell lines come with information on their growth medium. In most cases, the recommended medium and serum can be purchased from ATCC along with the cell line.
Use the following procedure to adapt a cell line to a new medium:
- Subculture the line at a 1:2 split ratio (split the culture in half) into two vessels.
- Maintain one with the original medium and continue to subculture these cells for the entire adaptation process. Use a 1:1 mix of the original and new medium in the second vessel. The culture grown in the original medium serves as a reference point as well as a safeguard in case the adapting cells do not survive the process. The low split ratio helps mitigate the stress associated with subculturing as well as with the new medium.
- Monitor cell growth in the two media and watch for any change in morphology or growth rate. If they are identical, subculture the adapting cells at the next passage with a 1:2 split ratio in a 1:3 medium mix (25% original, 75% new).
- Monitor the growth rate and morphology of the original and adapting cultures.
- At the next passage, split the adapting cultures 1:2 in a 1:7 medium mix (12.5% original, 87.5% new).
- Monitor the growth rate and morphology of the original and adapting cultures. If the cells are identical, then at the next passage split the adapting cells 1:2 in 100% new medium. At this point, the culture should be adapted to the new medium.
To confirm complete adaptation to the new medium, perform functional tests on cells derived from the original and new medium. If at any point in the process the adapting culture fails to perform as well as the reference culture, then allow the adapting culture more time and a few more passages in their current medium mix (eg, 1:3, 1:7, etc.) until they match the reference cells.
The same approach can be used to adapt cells to serum-free medium; simply decrease the serum level in the medium by half with each passage until a 0.06% (or lower) serum level is reached. At this point, the cells can be maintained in serum-free medium. If at any point the growth rate declines, then the serum level should be increased to the level where the cells grew normally. In this procedure, start with the “serum-free” medium supplemented with serum so that only the level of serum changes with each passage.
Temperature
Most animal cell lines require 37°C for optimum growth. Insect and amphibian cells require lower temperatures (such as 28°C) as do some animal cell lines which are temperature sensitive for their phenotypic characteristics. While cultured cells can withstand considerable drops in temperature and most can survive for several days at 4°C, few can tolerate even a few hours at more than 2°C above their optimal temperature. (See: NOTE 4)
NOTE 4
Regularly calibrate the temperature control system of incubators and use an alarm system when possible to warn against temperature increases above the optimum setting.
Examination of cultures
Observe the morphology and viability of cultures regularly and carefully. Examine the medium in the vessel for macroscopic evidence of microbial contamination. This includes unusual pH shifts (yellow or purple color from the phenol red), turbidity, or particles. Also, look for small fungal colonies that float at the medium-air interface. Specifically check around the edges of the vessel as these may not be readily visible through the microscope.
With an inverted microscope at low power (40×), check the medium for evidence of microbial contamination and the morphology of the cells. Bacterial contamination will appear as small, shimmering black dots within the spaces between the cells. Yeast contamination will appear as rounded or budding particles, while fungi will have thin filamentous mycelia. For nonadherent cells grown in flasks, such as hybridomas, this is a simple matter of viewing the flask directly on the microscope. For cells grown in spinner flasks or bioreactors, a sample of the cell suspension will need to be withdrawn and loaded into a microscope slide or hemocytometer for observation.
Most adherent cells should be attached firmly to the surface. In some cases, healthy cells will round up and detach somewhat during mitosis and appear very refractile. Following mitosis, they will reattach. Some of these will float free if the culture vessel is physically disturbed. In contrast, dead cells often round up and detach from the monolayer and appear smaller and darker (not refractile) than healthy cells.
Cells in suspension culture grow either as single cells or as clusters of cells. Viable cells appear round and refractile whereas dead cells appear smaller and darker. Occasionally, a portion of the cells will attach and grow on the side of the culture vessel and appear round or flattened. The percentage of attached cells varies with the culture conditions and the cell density. Cellular debris may also be observed in healthy cell populations. Some cell lines grow as mixed adherent and suspension cultures.
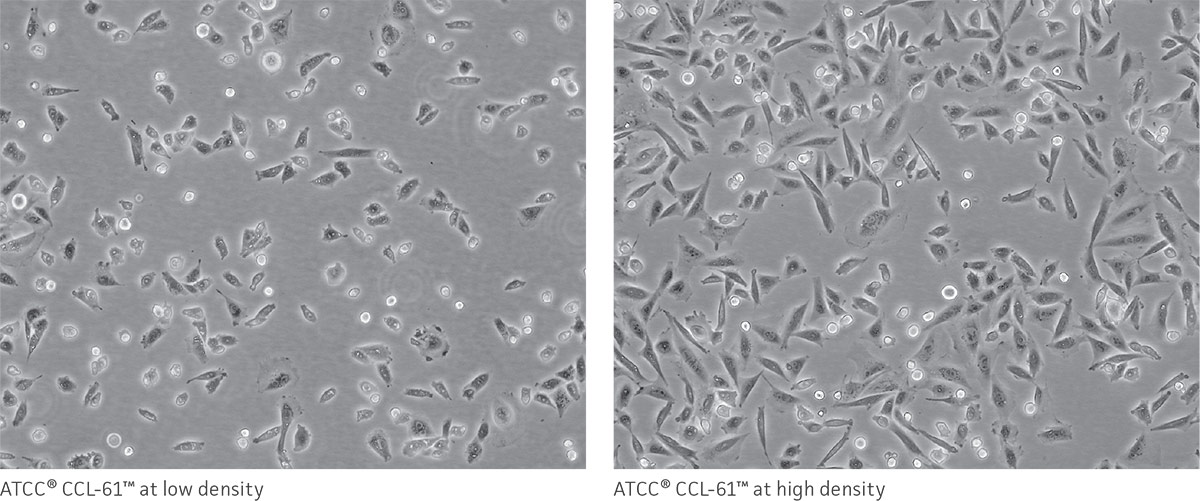
As a reference, photomicrographs for some ATCC cell lines are available on the website. Cells are shown at two different densities: just after subculturing (low) and just before they need to be subcultured (high).
In addition to daily examinations, periodically test a sample of the culture for the presence of fungi, bacteria, and mycoplasma. There are several methods that can be used to check for these contaminants. For additional information, refer to the section on microbial contamination.
Cell counting
Cell counts are necessary in order to establish or monitor growth rates as well as to set up new cultures with known cell numbers. Hemocytometers (also spelled hemacytometers) are commonly used to estimate cell number and determine cell viability. A hemocytometer is a fairly thick glass slide with two counting chambers, one on each side. Each counting chamber has a mirrored surface with a 3 × 3 mm grid of 9 counting squares. (See Figure 2.) The chambers have raised sides that will hold a coverslip exactly 0.1 mm above the chamber floor. Each of the 9 counting squares holds a volume of 0.0001 mL.
Hemocytometers are excellent for determining cell viability, but are not precise for determining cell number due to the relatively low number of cells actually counted. An automated counter will generate the most reliable data, particularly when used in combination with the viability data from a hemocytometer.
Count cells as follows:
- Clean, thoroughly dry, and assemble the hemocytometer with the cover slip.
- Transfer a small amount of cell suspension to the edge of each of the two counting chambers. Allow the cell suspension to be drawn into the counting chamber by capillary action.
- Place the hemocytometer under an inverted microscope and view the cells at 100× magnification.
- Focus on the quadrants, labeled 1, 2, 3, and 4 in Figure 2.
- Record the number of cells in each section. Average the number of cells, and multiply by the dilution factor. If the cells have not been diluted, this factor will be 104 cells/mL. Any dilution of the sample after it was removed from the cell suspension, such as using vital stain, needs to be included in the calculation.
For example, if the four counts are 60, 66, 69, and 75, the concentration would be 68 × 104 cells/mL for the sample that was loaded into the hemocytometer. For best results, adjust the concentration of the suspension so that 50 to 100 cells are in each of the four sections.
Most cultures will grow at an initial inoculum cell concentration ranging from 103 to 104 cells/cm2. Faster-growing cultures are usually set up at lower concentrations. Some cultures do not grow well unless a minimum concentration of cells is initially added; see the Product Sheet for details.
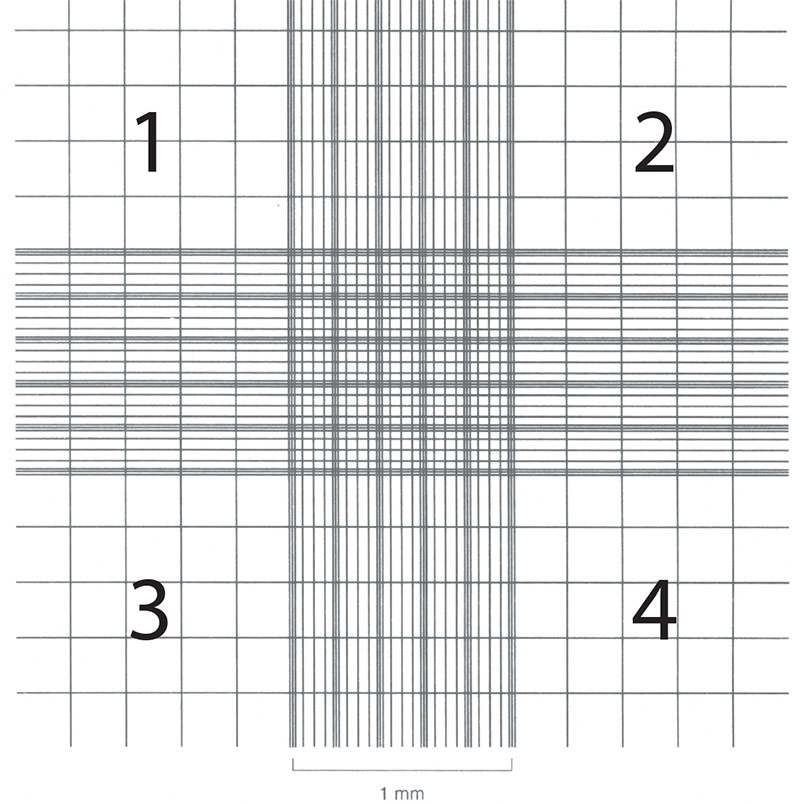
Figure 2: Hemocytometer grid with Neubauer ruling.
Cell viability
Viability assays measure the number of viable cells in a population. When combined with the total number of cells, the number of viable cells provides an accurate indication of the health of the cell culture. The most common and rapid methods rely upon the integrity of the cell membrane as an indicator of cell viability. Both trypan blue and erythrosin B stains are actively excluded by viable cells but are taken up and retained by dead cells, which lack an intact membrane.
While both stains are used in the same way, ATCC recommends erythrosin B in place of trypan blue for hematopoetic cells. When using trypan blue, incubate cells for two to five minutes prior to use. If not counted within this time, the cells will begin to deteriorate and take up the dye. Erythrosin B does not require an incubation period.
Erythrosin B stain generates more accurate results with fewer false negatives and false positives. Erythrosin B stain solution provides a clear background and does not bind serum proteins as avidly as trypan blue, making stained cells more distinct and easier to identify. Also, microbial contamination or precipitates in the cell culture are more readily apparent. Finally, trypan blue is toxic and a potential carcinogen.
For either stain use the following directions:
- Mix the cell suspension 1:1 with a 0.1% erythrosin B solution in PBS or 0.4% trypan blue solution in PBS.
- Load the cells in the erythrosin B solution directly into a clean, dry hemocytometer, but incubate the trypan blue solution for two to five minutes before loading.
- Nonviable cells will be stained red (erythrosin B) or dark blue (trypan blue). Cell viability is calculated as the number of unstained or viable cells divided by the total number of cells and expressed as a percentage.
Subculturing monolayer cells
Anchorage-dependent cell lines growing in monolayers need to be subcultured at regular intervals to maintain them in exponential growth. When the cells are near the end of exponential growth (roughly 70% to 90% confluent), they are ready to be subcultured. The subculturing procedure, including recommended split-ratios and medium replenishment (feeding) schedules, for each ATCC cell line is provided on the Product Information Sheet.
Subcultivation of monolayers involves the breakage of both intercellular and intracellular cell-to-surface bonds. For some cells that are loosely attached, a sharp blow with the palm of your hand against the side of the flask can dislodge them. Many require the digestion of their protein attachment bonds with proteolytic enzymes such as trypsin/EDTA. For some cell lines mechanical forces such as scraping to dislodge the cells is preferred. After the cells have been dissociated and dispersed into a single-cell suspension, they are diluted to the appropriate concentration and transferred into fresh culture vessels with the appropriate growth medium where they will reattach, grow and divide.
The procedure below is appropriate for most adherent cell lines. However, since every cell line is unique, incubation times and temperature, number of washes or the solution formulations may vary. In all cases, continually observe the cells with a microscope during the dissociation process to prevent damage by the dissociation solution. The amounts used in this procedure are for a 75-cm2 flask. Adjust volumes as appropriate for different sized vessels.
Monolayer subculturing
- Bring the trypsin-EDTA solution (ATCC 30-2101), balanced salt solution [Dulbecco’s Phosphate Buffered Saline without calcium or magnesium, ATCC 30-2200], and complete growth medium to the appropriate temperature for the cell line. In most cases, this is the temperature used to grow the cells (usually 37°C). For some sensitive cells, the trypsin-EDTA solution may need to be used at room temperature or 4°C.
- Remove and discard the cell culture medium from the flask.
- Rinse the cell monolayer with Dulbecco’s PBS without calcium or magnesium and remove.
- Add 2 mL to 3 mL of the trypsin-EDTA solution and incubate at the appropriate temperature. Check the progress of cell dissociation by microscopy. To avoid clumping, do not agitate the cells by hitting or shaking the flask while waiting for them to detach.
- Once the cells appear to be detached (5 to 15 minutes for most cell lines; they will appear rounded and refractile under the microscope), add 6 to 8 mL of complete growth medium with a pipette to the cell suspension to inactivate the trypsin. Gently wash any remaining cells from the growth surface of the flask. Check the cells with the microscope to be sure that most (>95%) are single cells. If cell clusters are apparent, continue to disperse the cells with gentle pipetting. (See: NOTE 5)
- Add 12 mL to 15 mL of fresh culture medium to a new flask and equilibrate this medium to the appropriate pH and temperature.
- Count the cells in suspension and determine their viability or simply divide them according to a routine split ratio and dispense them into the medium of the newly prepared flask. Do not add a concentrated cell suspension to an empty culture vessel as this can result in uneven cell attachment and growth.
- Place the flask back into the incubator. Examine the culture the following day to ensure the cells have reattached and are actively growing. Change the medium as needed; for most actively growing cultures two to three times per week is typical.
NOTE 5
For serum-free or low-serum medium, remove the trypsin-EDTA solution by gentle centrifugation (10 minutes at 125 × g) and then resuspend the cells in 6 mL to 8 mL of fresh medium. In some cases, the trypsin will need to be inactivated with a trypsin inhibitor.
Troubleshooting monolayer cell subculturing
Cells are difficult to remove.
- Inhibitors in the medium (such as serum) have inactivated the dissociating agents. Rinse the cell monolayer twice with Dulbecco’s PBS without calcium or magnesium before adding the dissociating solution. Or use the trypsin-EDTA solution in place of the Dulbecco’s PBS for the first rinse of the monolayer.
- The dissociating solution was too weak. Use higher enzyme concentrations, higher EDTA concentrations, or different and/or additional enzymes (eg, dispase, collagenase). Or incubate the cells at 37°C to increase the activity of the dissociating solution.
- The cells have been confluent for too long and the cell-to-cell junctions are so tight they prevented the dissociation agents from reaching the substrate-cell interface. In the future, subculture the cells before they become confluent.
Cells form clumps after dissociation.
- The dissociation procedure was too harsh and genomic DNA was released from lysed cells. Either the pipetting was too vigorous or the dissociating solution was too strong or too toxic (ie, the pH or osmolality of the buffer was incorrect). Add a drop of sterile DNAse (1 mg/mL in water) to the cell suspension to break down the DNA strands. In the future, treat the cells more gently during pipetting, shorten the incubation period, use a weaker dissociation solution (lower the enzyme concentration or remove the EDTA), or incubate at a lower temperature.
- The cells aggregated before dilution and dispersion into the medium. Hold the cell suspension on ice if there will be a delay between removing the cells from the flask growth surface and seeding a new flask.
- The cells were centrifuged too hard or too long when removing excess dissociation solution. Be sure to use gentle centrifugation (10 minutes at 125 × g).
Cells have difficulty reattaching to the flask.
- The dissociation procedure was too long and stripped away necessary attachment proteins from the cell membrane.
- Insufficient serum or attachment factors were present in the medium (common with serum-free medium). Add attachment factors to the medium and/or use a protein-coated flask (collagen, poly-L-lysine, fibronectin, gelatin, etc.).
- The dissociating solution was not inactivated or removed by centrifugation. Add additional serum or specific enzyme inhibitors (eg, soybean trypsin inhibitor) to the neutralizing medium or centrifuge (5 minutes at 125 × g) the cells down from the dissociation solution and resuspend in fresh medium.
Viability is lower than expected.
- The dissociating procedure was too harsh.
- The pH or osmolality of the balanced salt solution containing the dissociation agents is incorrect. Check these directly and/or use a fresh bottle.
- The dispersed cell suspension was left too long at too high a cell concentration prior to reseeding. Keep the cells on ice.
- The medium was faulty. Use the recommended formulation and make sure it contains all of the required additives.
Suspension cells
Most primary cultures, finite cell lines, and continuous cell lines are anchorage dependent and thus grow in monolayers attached to a surface. Other cells, particularly those derived from hematopoietic or certain tumor tissues, are anchorage independent and grow in suspension.
Cell propagation in suspension has several advantages over propagation in monolayer. Subculturing is a simple matter of dilution. There is little or no growth lag after splitting a suspension culture as there is with a monolayer culture, because there is none of the trauma associated with proteolytic enzyme dispersal. Suspension cultures require less lab space per cell yield, and scale-up is straightforward. Cells can be propagated in bioreactors similar to the fermentors used for yeast or bacteria cultures.
Depending upon the cell type, suspension cultures are seeded at densities from 2 × 104 to 5 × 105 viable cells/mL and can attain densities of 2 × 106 cell/mL. If cells are seeded at too low a density they will go through a lag phase of growth, grow very slowly, or die out completely. If cell densities are allowed to become too high, the cells may exhaust the nutrients in the medium and die abruptly. Recommended seeding and subculturing densities, media replenishment (feeding) schedules, and medium formulations for each ATCC cell line are provided on the Product Sheet as well as in the catalog description on the website.
Suspension cell subculturing
- Bring the complete growth medium to the appropriate temperature (usually 37°C) in a water bath.
- Thoroughly mix the cell/medium suspension; use a pipette to suspend cells grown in stationary flasks. Remove a small amount of the cell suspension to determine the cell density and viability using a hemocytometer and vital stain.
- Calculate the volume of cells required to re-seed the flask at the minimum density for that cell line, taking into consideration the amount of fresh medium that will be used.
- Add the appropriate volume of medium to the culture vessel and then add the cell suspension. Do not add the concentrated cell suspension to an empty flask. The same culture vessel can be reused, but the chances of contamination increase with each reseeding due to the buildup of small spills of medium on the flask opening.
- If necessary, “gas” the atmosphere of the flask with sterile-filtered CO2, seal the flask, and then incubate at the appropriate temperature.
It is generally not necessary to completely change the medium unless the cells attain a very high density or the medium has an acidic pH (yellow in color from the phenol red). To completely replace the medium, centrifuge the cells gently (10 minutes at 125 × g), decant the medium, and then resuspend the cells in fresh medium at the lower seeding density.
Troubleshooting suspension cell subculturing
Viability is lower than expected.
- The cell suspension was left too long at too high a cell concentration prior to subculture. In this case, the medium will have a low pH and be yellow in color. Completely change the medium by gently centrifuging the cells and resuspend in fresh medium at the lower seeding density.
- The cell suspension was diluted below the recommended cell density range. Recover the cells by centrifugation and resuspend in fresh medium at the appropriate cell density.
- The harvesting procedure was too harsh (pipetting too vigorous, cells were centrifuged too hard or too long, cells damaged during scraping or banging).
- The medium was faulty. Use the recommended formulation and make sure it contains all of the required additives.
Adapting a monolayer cell line to grow in suspension
Some cell lines such as L-929 (ATCC CCL-1), HeLa (ATCC CCL-2) and BHK-21 (ATCC CCL-10) can be adapted to grow in suspension. With time, a population of cells can be selected that does not self-aggregate or adhere to a growth surface as readily as the parental line. However, the newly selected line may have lost or acquired characteristics that are different from the original cell population. In most cases it will be necessary to maintain the culture in suspension with mechanical stirring. Keep in mind that most anchorage-dependent cells will grow in suspension only with the use of microcarrier beads.
The procedure below was developed for BHK-21 cells,4 but can be used as a starting point for most cell lines.
- Dissociate the cell monolayer using standard procedures. Centrifuge and resuspend the cell suspension in an appropriate spinner medium such as Joklik’s modified Eagle’s Minimum Essential Medium (EMEM). Spinner media have reduced levels of calcium and magnesium.
- Count the cell suspension, and then seed two or more spinner flasks with 5 × 105 cells/mL. This density may need to be adjusted for your particular cell line. The sides of the culture flask may need to be siliconized to prevent the cells from sticking to the glass.
- Observe the cultures daily. Remove samples and record the number of viable cells for each flask.
- Every three days, collect the cells growing in suspension by centrifugation (10 minutes at 125 × g). Count, and re-seed a fresh flask with fresh medium at 2.5 × 105 cells/mL. Depending on how well (or not) the cells adapt to growth in suspension, they may need to be combined with cells from different flasks to achieve the necessary cell density.
- If there is a significant amount of cells attached to the walls of the culture vessel, particularly at the surface of the medium, remove them with trypsin-EDTA and discard them. If the cells in suspension are badly clumped, they can be dispersed with the trypsin-EDTA solution, collected by centrifugation, and then re-seeded into the flask as the appropriate density. This treatment may be necessary for the first few subcultures.
- Continue to monitor the cells and subculture them every three days. Over time, they should adapt to growth in suspension and attain a constant growth rate.
A complete growth medium consists of a basal cell culture medium supplemented with ingredients such as sera, growth factors, trace elements, and hormones. There are numerous formulations ranging from simple, basic mixtures containing the minimum requirements for growing many cell lines to complex serum-free mixtures specific for growing a single fastidious cell line. The choice of a medium for a particular cell line is somewhat empirical. Many medium formulations are available commercially in powder or liquid form. (See: NOTE 6)
ATCC lists complete medium formulations, plus all handling and passage information, for all ATCC cell lines both in the online catalog description and on the Product Sheet. Additionally, ATCC offers a full line of media, sera, and reagents for culturing cells. These are the same reagents used at ATCC for cell growth and propagation. See descriptions of ATCC cell culture products.
NOTE 6
Formulations can vary widely among suppliers, even for media with similar or identical names. Be sure to read catalog descriptions, formulations, and medium labels carefully to ensure that the appropriate medium is used. For best results start cell cultures in the same medium used and distributed by ATCC (listed on the Product Sheet).

Complete growth media
Cell culture media
Cell culture media are complex mixtures of salts, carbohydrates, vitamins, amino acids, metabolic precursors, growth factors, hormones, and trace elements. The requirements for these components vary among cell lines, and these differences are partly responsible for the extensive number of medium formulations. Carbohydrates are supplied primarily in the form of glucose. In some instances, glucose is replaced with galactose to decrease lactic acid build-up, as galactose is metabolized at a slower rate. Other carbon sources include amino acids (particularly L-glutamine) and pyruvate.
In addition to nutrients, the medium helps maintain the pH and osmolality in a culture system. The pH is maintained by one or more buffering systems; CO2/sodium bicarbonate, phosphate, and HEPES are the most common. Sera will also buffer a complete medium. Phenol red, a pH indicator, is added to medium to colorimetrically monitor changes in pH. Commonly used culture media include the following:
Eagle’s Minimum Essential Medium (EMEM) was among the first widely used media and was formulated by Harry Eagle from his earlier and simpler basal medium (BME). BME was developed for culturing mouse L cells (ATCC CCL-1) and HeLa cells (ATCC CCL-2). Over time, there have been numerous variations on the EMEM formula for different applications. ATCC EMEM (ATCC 30-2003) contains Earle’s balanced salt solution, nonessential amino acids, and sodium pyruvate. It is formulated with a reduced sodium bicarbonate concentration (1,500 mg/l) for use with 5% CO2 (see Sodium Bicarbonate and Buffering). Because EMEM is a simple medium, it is often fortified with additional supplements or higher levels of serum.
Dulbecco’s Modified Eagle’s Medium (DMEM) has roughly twice the concentration of amino acids and four times the amount of vitamins as EMEM, as well as ferric nitrate, sodium pyruvate, and some supplementary amino acids (though not all nonessential amino acids). The original formulation contained 1,000 mg/L of glucose, but in the more commonly used variations this amount was increased to 4,500 mg/L.
ATCC DMEM (ATCC 30-2002) has 4,500 mg/L of glucose and a reduced sodium bicarbonate concentration (1,500 mg/L) for use with 5% CO2.
Iscove’s Modified Dulbecco’s Medium (IMDM) was formulated for growth of lymphocytes and hybridomas. Compared to DMEM, it has additional amino acids, vitamins and inorganic salts. Potassium nitrate was substituted for ferric nitrate. It also contains HEPES and selenium. ATCC IMDM (ATCC 30-2005) has a reduced sodium bicarbonate concentration (1,500 mg/L) for use with 5% CO2.
Hybri-Care Medium (ATCC 46-X) is a combination and modification of DMEM and NCTC 135 medium supplemented with insulin, oxalacetic acid, and HEPES. It is based on the formulation used by David H. Sachs and collaborators5 for the propagation of hybridomas and other fastidious cell lines.
McCoy’s 5A and RPMI-1640 were developed at Roswell Park Memorial Institute (RPMI) in Buffalo, New York. McCoy’s 5A (ATCC 30-2007) was originally used to grow Novikoff hepatoma cells and will support the growth of primary cultures.
RPMI-1640 is a modification of McCoy’s 5A and was developed for the long-term culture of peripheral blood lymphocytes. RPMI-1640 will support the growth of a wide variety of cells in suspension as well as a number of cells grown as monolayers.
ATCC RPMI-1640 (ATCC 30-2001) was modified to contain higher amounts of glucose (4,500 mg/L), sodium pyruvate, and HEPES buffer. It also contains a reduced concentration of sodium bicarbonate (1,500 mg/L) for use with 5% CO2.
Ham’s Nutrient Mixtures were originally developed to support the clonal outgrowth of Chinese hamster ovary (CHO) cells (ATCC CCL-61). As with EMEM, there have been numerous modifications to the original formulation including Ham’s F-12 medium, a more complex formulation than the original F-10 suitable for serum-free propagation.
Kaighn’s modification of Ham’s F-12 (Ham’s F-12K) was designed to support the growth and differentiation of primary cells with or without serum. F-12K has increased amounts of amino acids, pyruvate, biotin, calcium, magnesium, putrescine, and phenol red in addition to other modifications from the F-12 formula. ATCC Ham’s F-12K (ATCC 30-2004) has a reduced sodium bicarbonate concentration (1,500 mg/L) for use with 5% CO2.
DMEM/F12 Medium is a 1:1 mixture of Dulbecco’s modified EMEM and Ham’s F-12. It is an extremely rich and complex medium and will support the growth of a broad range of cell types in both serum and serum-free formulations.
ATCC DMEM/F12 medium (ATCC 30-2006) has a reduced sodium bicarbonate concentration (1,500 mg/L) for use with 5% CO2.
Leibovitz’s L-15 Medium (ATCC 30-2008) is formulated for use without CO2 incubation as is found in teaching laboratories or when collecting biopsy samples. The standard sodium bicarbonate/CO2 buffering system is replaced by a combination of phosphate buffers, free-base amino acids, higher levels of sodium pyruvate, and galactose. Cell cultures can be grown in CO2 incubators with L-15 medium provided there is no exchange between the air in the culture vessel with that of the incubator (ie, caps of flasks are tightly closed).
Media formulations
Formulations of media available from ATCC can be found online. Please note that there are cell lines in the collection that require media not currently sold by ATCC.
Media ingredients
Sodium bicarbonate and buffering
Cells produce and require small amounts of carbon dioxide for growth and survival.6 In culture media, dissolved CO2 is in equilibrium with bicarbonate ions and many medium formulations take advantage of this CO2/bicarbonate reaction to buffer the pH of the medium. CO2 dissolves freely into the medium and reacts with water to form carbonic acid. As the cells metabolize and produce more CO2, the pH of the medium decreases as the chemical reaction below is driven to the right:
H2O + CO2 ↔ H2CO3 ↔ H+ + HCO3-
The optimal pH range of 7.2 to 7.4 can be maintained by supplementing the medium with sodium bicarbonate and regulating the level of CO2 in the atmosphere above the medium as shown by the reaction below:
H2O + CO2 + NaHCO3 ↔ H+ + Na+ + 2HCO3-
In tissue culture, cells are grown either in open systems (where there is free exchange of the atmosphere immediately above the medium with the atmosphere of the incubator) or in closed systems (where the two atmospheres are kept separate). The buffering system employed in the medium needs to be matched to the culture system. Otherwise the cells may be subject to metabolic stress which will impair their performance.
In closed systems the level of CO2 is regulated by the metabolism of the cells. The culture vessel must be sealed (flasks tightly capped) to retain any CO2 generated by the cells. Consequently, closed systems provide additional protection against contamination and have simpler incubator requirements than open systems. Closed systems usually require media with buffers based on Hanks’ balanced salt solution having relatively low levels of sodium bicarbonate.
In open systems, humidity (to reduce evaporation) and a means of regulating CO2 levels (if the culture medium contains sodium bicarbonate) are required during incubation to maintain the pH of the culture medium. Open systems usually require the higher levels of sodium bicarbonate found in Earle’s salt solution combined with a 5 to 10% CO2 atmosphere supplied by the incubator. In general, 1.2 g/L to 2.2 g/L of sodium bicarbonate is used with 5% CO2 whereas 3.7 g/L sodium bicarbonate is used with 10% CO2. The exact amount will depend upon the medium formulation.
In some cases, researchers “gas” the atmosphere of the culture vessel with a stream of sterile-filtered 5% CO2/95% air mixture and then tightly seal the flask prior to incubation in a nonhumidified and non-CO2 incubator.7 While these culture vessels work with simpler non-humidified, non-CO2 incubators, the medium requirements are those of an open system.
All ATCC media, with the exception of Leibovitz’s L-15 (ATCC 30-2008), are designed to be used with 5% CO2 levels. Most have a sodium bicarbonate concentration of 1.5 g/L and are supplemented with extra sodium pyruvate. ATCC modification of McCoy’s 5A (ATCC 30-2007) has a slightly higher levels of sodium bicarbonate (2.2 g/L) and does not contain sodium pyruvate.
While most commercial formulations of liquid media do contain the appropriate amount of sodium bicarbonate, it is generally omitted from the powdered form and needs to be added before use. Some medium formulations incorporate other buffering systems such as phosphate or HEPES in addition to CO2/sodium bicarbonate. These media have the advantage of maintaining optimal pH in an open system when the culture vessel is removed from the enriched CO2 atmosphere of the incubator.
HEPES buffer
HEPES and other organic buffers can be used with many cell lines to effectively buffer the pH of the medium.8 Indeed, some standard medium formulations include HEPES. However, this compound can be toxic, especially for some differentiated cell types, so evaluate its effects before use.9 HEPES has been shown to greatly increase the sensitivity of media to the phototoxic effects induced by exposure to fluorescent light.10,11
Phenol red
Phenol red is used to monitor the pH of media. During cell growth, the medium changes color as it changes pH due to metabolites released by the cells. At low pH levels, phenol red turns the medium yellow, while at higher pH levels it turns the medium purple. For most tissue culture work (pH 7.4), the medium should be bright red.
Unfortunately, phenol red can mimic the action of some steroid hormones, particularly estrogen. For studies with estrogen-sensitive cells, such as mammary tissue, use media without phenol red. Additionally, the sodium-potassium ion homeostasis is upset when phenol red is included in some serum-free formulations; this effect is neutralized by the inclusion of serum or bovine pituitary hormone in the medium.12 Phenol red is frequently omitted from studies with flow cytometry as its color interferes with detection.
L-Glutamine
L-Glutamine (ATCC 30-2214) is an essential amino acid required by virtually all mammalian and insect cells grown in culture. It is used for protein production, as an energy source, and in nucleic acid metabolism. It is also more labile in liquid cell culture media than other amino acids. The rate and extent of L-glutamine degradation are related to storage temperatures, age of the product, and pH.
Because L-glutamine is so labile, it is often omitted from commercial liquid medium preparations to lengthen the product shelf life. In these cases, it must be aseptically added prior to use. L-Glutamine is not as labile in dry form and most powdered medium formulations do include it.
In some cases, the addition of L-glutamine to complete cell culture medium can extend the usable life of the medium. If L-glutamine is suspected to be a limiting factor during cell culture, a simple test of ‘spiking’ the medium with a small amount of L-glutamine will determine whether or not more is required. Simply add a small amount of L-glutamine (~2 mM final concentration) to the culture medium. If the cell growth rate increases, L-glutamine is most likely deficient and more should be added. Alternately, the concentration of L-glutamine can be measured directly by standard analytical means such as HPLC (High Performance Liquid Chromatography).
L-Glutamine concentrations for mammalian cell culture media can vary from 0.68 mM in Medium 199 to 4 mM in Dulbecco’s Modified Eagle’s Medium. Invertebrate cell culture media, such as Schneider’s Drosophila medium, may contain as much as 12.3 mM L-glutamine.
Use caution when adding more L-glutamine than is called for in the original medium formulation. L-Glutamine degradation results in the build-up of ammonia which can have a deleterious effect on some cell lines. For most cell lines, ammonia toxicity is more critical for cell viability than L-glutamine limitation.
Nonessential amino acids
All medium formulations contain the ten essential amino acids as well as cysteine, glutamine, and tyrosine. The inclusion of the other non-essential amino acids (alanine, asparagine, aspartic acid, glycine, glutamic acid, proline, and serine) in some media formulations reduces the metabolic burden on the cells allowing for an increase in cellular proliferation.
Sodium pyruvate
Pyruvate is an intermediary organic acid metabolite in glycolysis and the first component of the Embden-Meyerhof pathway. It can pass readily into or out of the cell. Its addition to tissue culture medium provides both an energy source and a carbon skeleton for anabolic processes. Pyruvate may help in maintaining certain specialized cells, in clonal selection, in reducing the serum concentration of the medium,7 and in reducing fluorescent light-induced phototoxicity.10 Cellular metabolism of pyruvate produces carbon dioxide which is given off into the atmosphere and becomes bicarbonate in the medium. Sodium pyruvate is added to give a final concentration of 1 mM in most media, but is increased to 5 mM in Leibovitz’s L-15 medium primarily to facilitate use in CO2-free environments.
Media supplements
The complete growth media recommended for some cell lines requires the addition of components not already available in the base media and serum. These components include hormones, growth factors and signaling substances that sustain proliferation and maintain normal cell metabolism.
Supplements are usually prepared as 100× (or higher) stock solutions in serum-free medium. Some supplements may need to be dissolved in a solvent prior to subsequent dilution in serum-free medium to the stock concentration. Stock concentrations should be aliquoted into small volumes and stored at an appropriate temperature; most stock concentrations can be stored at –80°C, but check with your supplier prior to storing.
The addition of supplements can change the final osmolality of the complete growth medium, which may have a negative effect on the growth of cells in culture. It is best to recheck the osmolality of the complete growth medium after small volumes of supplement stock solutions are added; optimal osmolality for most vertebrate cell lines should fall between 260 mOSM/kg and 320 mOSM/kg.
After supplements have been added to a base medium, the shelf life of the complete growth medium should be determined on a case-by-case basis. Complete media containing protein supplements (eg, epidermal growth factor, bovine serum albumin, etc.) tend to degrade faster than base media alone. Most complete growth media can be stored in aliquots at 2°C to 8°C for about a month. However, if any supplement is expected to expire before the one-month period has passed, the expiration date for the complete growth media should follow suit. Some fastidious cell lines may require that components be added immediately before use. Do not freeze complete growth medium. Freezing cell culture media at –70°C or below causes some of the growth factors and/or vitamins to precipitate out of solution. It can be very difficult to get these components to go back into solution after thawing, even if warmed to 37°C. ATCC recommends storing media between 2°C and 8°C, away from light.
For additional information regarding the preparation, storage, or usage of specific supplements, contact your local supplier or consult with the manufacturer’s Product Information Sheet.
Osmolality
The osmolality of cell culture media for most vertebrate cells is kept within a narrow range from 260 mOsm/kg to 320 mOsm/kg, even though most established cell lines will tolerate a rather large variation in osmotic pressure. In contrast, the osmolality requirements for some invertebrate cell lines fall outside of this range. For example, the snail embryo requires medium of about 155 mOsm/kg, while some insect cells prefer 360 mOsm/kg to 375 mOsm/kg. Most commercially available liquid media report osmolality and it is advisable to check the osmolality of any medium after the addition of saline solutions, drugs or hormones dissolved in an acid or base solution, or large volumes of buffers (eg, HEPES).
Antibiotics and antimycotics
Antibiotics and/or antimycotic agents are added to cell culture media as a prophylactic to prevent contamination, as a cure once contamination is found, to induce the expression of recombinant proteins, or to maintain selective pressure on transfected cells.
Routine use of antibiotics or antimycotics for cell culture is not recommended unless they are specifically required, such as G418 for maintaining selective pressure on transfected cells. Antibiotics can mask contamination by mycoplasma and resistant bacteria. Further, they can interfere with the metabolism of sensitive cells. Avoid antimycotics as they can be toxic to many cell lines.
While cell lines can be cured of microbial contamination with antibiotics and/or antimycotics, this is not recommend unless the cell line is irreplaceable; the process is lengthy and there is no guarantee contamination will be eliminated. Even if the contamination is eliminated, there is no way of ensuring that the resulting cell line will have the same characteristics as the initial one due to the stress of the treatment. It is best to discard the cell line and start over with new stocks. Mycoplasma contamination in particular is very difficult to eliminate. In some cases, antibiotic use for short periods of time can serve as a valuable prophylactic. For example, antibiotic use is recommended when developing and working with primary culture and when using flow cytometry to isolate subpopulations.
If an antibiotic is used in medium, penicillin-streptomycin solution (ATCC 30-2300) can be added at 0.5 to 1 mL of solution per 100 mL of cell culture medium for a final concentration of 50 to 100 IU/mL penicillin and 50 to 100 µg/mL streptomycin. Gentamicin sulfate, another antibiotic, is used at 50 to 100 µg/mL. The antimycotic amphotericin B is used at 2.5 µg/mL.13 These concentrations apply to media that contain serum. For serum-free media, reduce the concentrations by at least 50%.
Animal sera
Sera serve as a source for amino acids, proteins, vitamins (particularly fat-soluble vitamins such as A, D, E, and K), carbohydrates, lipids, hormones, growth factors, minerals, and trace elements. Additionally, serum buffers the culture medium, inactivates proteolytic enzymes, increases medium viscosity (which reduces shear stress during pipetting or stirring), and conditions the growth surface of the culture vessel. The exact composition is unknown and varies from lot to lot, although lot-to-lot consistency has improved in recent years.
Sera from fetal and calf bovine sources are commonly used to support the growth of cells in culture. Fetal serum is a rich source of growth factors and is appropriate for cell cloning and for the growth of fastidious cells. Calf serum, because of its lower growth-promoting properties, is used in contact-inhibition studies with NIH/3T3 cells (ATCC CRL-1658). In contrast to fetal or calf sera, horse serum is collected from a closed herd of adult animals ensuring lot-to-lot consistency. Horse serum is less likely to carry the contaminants found in bovine sera such as viruses and less likely to metabolize polyamines which may be mitogenic for some cells. Horse and bovine calf sera are less expensive and more readily available than fetal bovine serum. The pricing and availability of fetal serum fluctuates considerably.
Unfortunately, naturally derived products from bovine sources may contain adventitious viruses such as bovine viral diarrhea virus (BVDV), bovine parvovirus, bovine adenovirus, and blue tongue virus. All reputable suppliers test their products for infectious virus by several methods including fluorescent antibody, cytopathic effect, and hemadsorption. These products are also screened for the standard microbial contaminants such as bacteria, fungi, and mycoplasma.
BVDV, in contrast to the other virus contaminants, is present in nearly all bovine serum at very low levels even when tests for infectious virus are negative. Fortunately, very few cell lines (except those of bovine origin) are susceptible to this virus. For the few sensitive cell lines, use non-bovine sera or irradiated bovine sera. Several ATCC cell lines were tested for BVDV contamination14 and the results of this study are indicated in the cell line description on the website. Bovine-derived products also may contain the agent responsible for bovine spongiform encephalopathy (BSE). Unfortunately, there is no test for the presence of this agent and we highly recommend that you obtain all bovine products (including sera) from countries not affected by BSE such as the United States, Australia, and New Zealand.
At one time animal serum was a major source of mycoplasma contamination of tissue culture cells. However, nearly all sera today are filtered through several 0.1-µm pore (or smaller) filters which effectively remove this organism.
ATCC offers the following three types of animal sera:
- Fetal Bovine Serum (also known as fetal calf) — ATCC 30-2020
- Fetal Bovine Serum qualified for embryonic stem cells — ATCC SCRR-30-2020
- Iron-supplemented Calf Bovine Serum — ATCC 30-2030
These products are rigorously tested for adventitious infective agents and sourced from only U.S. herds. Further, each lot is tested for its ability to support cell growth and is the same sera used in ATCC labs.
Storage
Store sera at −20°C or colder for storage over 30 days. ATCC sera are routinely stored at −70°C. Do not store sera at temperatures above −20°C for any length of time. Avoid repeated freeze-thaws by dispensing and storing in aliquots.
Thawing
The following procedure is used to thaw serum:
- Place frozen serum in a refrigerator at 2°C to 8°C overnight.
- Put the bottles in a 37°C water bath and gently agitate from time to time to mix the solutes that tend to concentrate at the bottom of the bottle.
Do not keep the serum at 37°C any longer than necessary to thaw it, and do not thaw the serum at higher temperatures. Thawing serum in a bath above 40°C without mixing may lead to the formation of a precipitate inside the bottle.
Turbidity and precipitates
All sera may retain some fibrinogen. Because external factors may initiate the conversion of fibrinogen to fibrin, flocculent material or turbidity may be observed after serum is thawed. The presence of this material does not alter the serum’s performance. If the presence of flocculent material or turbidity is a concern, it can be removed by filtration through a 0.45-µm filter.
A precipitate can form in serum when incubated at 37°C or higher for prolonged periods of time which may be mistaken for microbial contamination. This precipitate may include crystals of calcium phosphate, but does not alter the performance of the serum as a supplement for cell culture. Heat inactivation of sera can also cause the formation of precipitates.
Heat inactivation
ATCC does not routinely use heat-inactivated serum unless specifically required for a particular cell line. Heat inactivation is usually unnecessary and can be detrimental to the growth of some cells. It will reduce or destroy growth factors present in the serum.
Heat inactivation was originally performed to inactivate complement (a group of proteins present in sera that are part of the immune response) as well as to destroy mycoplasma contaminants. Today, mycoplasma contamination, if any, is removed by filtration. Removal of complement is usually unnecessary, but can be important when preparing or assaying viruses or in cytotoxicity tests. According to a study by HyClone,15 warming serum to 37°C inactivates heat-labile complement factors. A few types of cell lines grow better in heat-inactivated sera such as embryonic stem cells16 and many insect cell lines.17
The following procedure can be used to heat-inactivate serum:
- Thaw serum.
- Preheat a water bath to 56°C. Use sufficient water to immerse the bottle above the level of serum.
- Mix thawed serum by gentle inversion and place in the 56°C bath. The temperature of the water bath will drop.
- When the temperature of the water bath reaches 56°C again, continue to heat for an additional 30 minutes. Mix gently every 5 minutes to insure uniform heating.
- Remove serum from water bath, cool quickly (slow cooling can sometimes reverse the inactivation of complement activity), and store at −20°C or colder.

Culture vessels and surfaces
Vessels
Culture vessels provide a contamination barrier to protect the cultures from the external environment while maintaining the proper internal environment. For anchorage-dependent cells, the vessels provide a suitable and consistent substrate for cell attachment. Other characteristics of vessels include easy access to the cultures and optically clear viewing surfaces.18
Originally all culture vessels were glass. Drawbacks for glass include the heavy weight, expense, labor-intensive cleaning, and poor microscopic viewing compared to plastic. By the 1960s, surface treatment techniques were developed for polystyrene, allowing plastic vessels to replace glass for most cell culture applications.
The information below focuses on standard culture vessels used by many researchers. Large-scale culture equipment is not included.
Selecting the right vessel
First, match the characteristics of the cells to be grown with the characteristics of the different culturing systems. There are three basic types of cell cultures:
- Anchorage dependent, which must become attached to a surface to grow (for example, human diploid fibroblasts).
- Anchorage independent, which grow in suspension (most blood-derived cell cultures).
- Cells that can grow either attached or in suspension (many transformed cell lines such as HeLa and BHK-21).
Understand the growth requirements of the cultures to help select the best culture system. There are four basic culture systems:
- Stationary monolayer cultures which are grown in undisturbed flasks, dishes, and multiwell plates. These are the easiest culture systems to use and require the least amount of equipment. However, these systems are very labor intensive for producing large quantities of cells.
- Moving monolayer cultures which are grown primarily in roller bottles. These vessels are slowly rotated (approximately 0.5 rpm to 1 rpm) on motorized racks or drums and are widely used for producing large quantities of cells. Roller bottles employ simple technology but require an investment in the appropriate equipment.
- Stationary suspension cultures which are grown without agitation in untreated dishes and flasks. These are best for growing small volumes of anchorage-independent cells that grow poorly in traditional stirred suspension cultures.
- Moving suspension cultures which are grown in mechanically stirred vessels (spinner flasks), bioreactors, or fermentors. These systems are the most economical in terms of space, labor and media; as a result, stirred suspension cultures are usually the method of choice for producing large volumes of cells both in the lab and in industry. Many anchorage-dependent cells can be adapted to grow on microcarriers to take advantage of these systems.
Next, decide whether the cells will be grown as an open system or as a closed system (see the section on sodium bicarbonate). Open-system plastic dishes are less expensive than closed-system flasks, but require more expensive incubators that can regulate the CO2 and humidity in the atmosphere. Closed systems provide additional protection against contamination and have simpler incubator requirements. All dishes and multiwell plates are open systems. All other culture vessels can be used in either mode by leaving caps loose for an open system or tightened for a closed system. The plastic walls of culture vessels are slightly permeable to carbon dioxide and oxygen, permitting a very small amount of gas exchange. This is not a problem in most culture applications, but may interfere with anoxia experiments or long-term storage of media.19 Caps that allow gas exchange when the cap is fully tightened are available to reduce opportunities for flask spills and contamination in open systems.
The last step is matching the desired cell yield with an appropriately sized culture vessel. For monolayer cultures, the yield is limited by the area of treated growth surface. Approximately 0.5 × 105 cells/cm2 to 1 × 105 cells/cm2 of treated surface is a typical yield for confluent continuous mammalian cell lines. For suspension cultures the total cell yield is determined by the working volume of the vessel. In stirred systems, cell concentrations can easily reach between 1 × 106 cells/mL and 2 × 106 cells/mL of medium. However, the exact yields will need to be determined empirically for each cell line. ATCC strongly recommends that cells be maintained in the logarithmic phase of growth, and not be allowed to enter the stationary phase. Anchorage-dependent cell lines are routinely passaged or split before they reach confluency.
Flasks
Alexis Carrel developed the first glass flasks in the 1920s. Harry Earle developed the more traditional straight neck rectangular (also hexagonal) glass T-flasks in the 1940s. Today, plastic flasks are available with a range of growing areas, a variety of shapes, with several different neck designs. Choice of design depends on the cell culture techniques used as well as personal preference. The more common sizes are listed below.
| Description | Growth area (cm2) | Recommended working volume (mL) | Cell yield* |
|---|---|---|---|
| T-25 | 25 | 5 to 10 | 2.5 × 106 |
| T-75 | 75 | 15 to 25 | 7.5 × 106 |
| T-150 | 150 | 30 to 50 | 15.0 × 106 |
| T-175 | 175 | 35 to 60 | 17.5 × 106 |
| T-225 | 225 | 45 to 75 | 22.5 × 106 |
*Cell line dependent. Based upon a density of 1 × 105 cells/cm2.
Cell culture dishes
Cell culture dishes offer the best economy and access to the growth surface. This makes them the vessels of choice for cloning or other manipulations such as scraping that require direct access to the cell monolayer. They must be used with incubators that control CO2 and humidity. Most manufacturers offer dishes in four diameters: 35 mm, 60 mm, 100 mm, and 150 mm. These are nominal diameters and may not be the actual diameter of the growth surface. Cell culture dishes are available with either specially treated surfaces for growing anchorage-dependent cells, or untreated (native) surfaces for growing suspension cultures where attachment is not desired.
| Description | Growth area (cm2) | Working volume (mL) | Cell yield* |
|---|---|---|---|
| 35 | 8 | 1 to 2 | 0.8 × 106 |
| 60 | 21 | 4 to 5 | 2.1 × 106 |
| 100 | 55 | 10 to 12 | 5.5 × 106 |
| 150 | 148 | 28 to 32 | 14.8 × 106 |
*Cell line dependent. Based upon a density of 1 × 105 cells/cm2.
Multiwell plates
These widely used vessels were originally designed for virus titration, but have since become popular in many other applications, especially hybridoma production, high-throughput screening, and toxicity testing. Multiwell plates offer significant savings in space, media, and reagents when compared to an equal number of dishes. They are more convenient to handle, especially if the pipettors, plate washers, readers, and other equipment for processing these plates are used. They must be used with incubators that control humidity and CO2 levels.
| Description | Growth well (cm2) | Working volume/ well (mL) | Cell yield* |
|---|---|---|---|
| 96-well | 0.32 | 0.1 to 0.2 | 0.32 × 105 |
| 48-well | 1.00 | 0.3 to 0.6 | 0.8 × 105 |
| 24-well | 1.88 | 0.5 to 1.2 | 1.9 × 105 |
| 12-well | 3.83 | 1.0 to 2.4 | 3.8 × 105 |
| 6-well | 9.40 | 2.0 to 3.0 | 9.5 × 105 |
*Cell line dependent. Based upon a density of 1 × 105 cells/cm2.
Roller bottles
The roller bottle was developed for cultivating large numbers of anchorage-dependent cells.20 Today they provide a more economical means for cultivating large volumes of cells using essentially the same culture techniques as with flasks but with considerably less labor. Besides the traditional smooth wall design, roller bottles are available with small ridges that approximately double the surface area available for growing cells without increasing the dimensions of the bottles.
| Description | Growth area (cm2) | Working volume (mL) | Cell yield* |
|---|---|---|---|
| Small | 490 | 100 to 150 | 4.9 × 107 |
| Standard | 850 | 170 to 250 | 8.5 × 107 |
| Pharmaceutical | 1750 | 340 to 500 | 17.5 × 107 |
*Cell line dependent. Based upon a density of 1 × 105 cells/cm2.
Surface coatings and feeder cells
Most tissue culture work uses disposable polystyrene vessels. The vessel surface is treated to render it hydrophilic (wettable). Most cell lines in the ATCC collection are cultivated on treated plastic surfaces in dishes, flasks, or roller bottles. Since the properties of tissue culture plastic can vary among manufacturers, samples should be evaluated for their ability to support cell growth and propagation prior to use. ATCC routinely uses the SelecT fully automated cell culture system. Some fastidious cell lines require further treatment of the growth surface before they will attach and proliferate. The most common techniques include coating the surface with serum, collagen, laminin, gelatin (ATCC PCS-999-027), poly-L-lysine, or fibronectin.
Beyond simple attachment, some cells require specialized surface treatment in order for them to differentiate into more tissue-like formations. For example, endothelial cells will form tubules and neuronal cells will extend neurite processes when cultured on a surface of extracellular matrix (ECM) proteins. These ECM proteins closely resemble the basal lamina membrane surrounding cells in tissue and not only provide attachment points, but modulate signal transduction from external growth factors and hormones, influence the permeability of ions and nutrients, and actively “communicate” with intracellular processes through integrins.
Finally, some cells, particularly when seeded at low densities as for cloning, require the support of living cells. Most cells are “happier in a crowd.” Feeder layer cells supply a crowd by conditioning the medium through metabolic leakage and/or the active secretion of growth and other factors. They also provide a support matrix for cell attachment and proliferation. To prevent feeder layer cells from overgrowing the cells of interest, they are treated to prevent division. Common methods include irradiation with X-rays or gamma rays or treatment with mitomycin C. Each of these treatments damages cellular DNA so that the cells continue to metabolize but can no longer proliferate. ATCC offers a variety of well-characterized feeder cells.
| ATCC number | Product name | Description |
|---|---|---|
| CRL-2581 | C166 | Mouse embryonic endothelial cells |
| CRL-2583 | C166–GFP | Mouse embryonic endothelial cells with GFP expression |
| CRL-2749 | OP9 | Mouse embryonic bone marrow stromal cells |
| 55-X | IRR-MRC-5 | Irradiated MRC-5 cells (human diploid lung fibroblast) |
| SCRC-1007 | AFT024 | Mouse embryonic liver fibroblasts |
| SCRC-1007.1 | Irradiated AFT024 | Irradiated mouse embryonic liver fibroblasts |
| SCRC-1008 | MEF (C57BL/6) | Mouse embryonic fibroblasts |
| SCRC-1008.1 | MEF (C57BL/6) IRR | Irradiated mouse embryonic fibroblasts |
| SCRC-1040 | MEF (CF-1) | Mouse embryonic fibroblasts |
| SCRC-1040.1 | MEF (CF-1) IRR | Irradiated mouse embryonic fibroblasts |
| SCRC-1040.2a | MEF (CF-1) MITC | Mitomycin C treated mouse embryonic fibroblasts |
| SCRC-1041 | HFF-1 | Human foreskin fibroblasts |
| SCRC-1041.1 | HFF-1 IRR | Irradiated human foreskin fibroblasts |
| SCRC-1045 | MEF (DR4) | Multidrug resistant mouse fibroblasts |
| SCRC-1049 | SNL76/7 | STO fibroblasts with G418 resistance and endogenous expression of LIF |
| SCRC-1050 | SNLP 76/7-4 | STO fibroblasts with resistance to G418 and puromycin plus endogenous expression of LIF |

Cryopreservation
Most cell cultures can be stored for many years, if not indefinitely, at temperatures below –130°C (cryopreservation). ATCC has recovered cells from cultures cryopreserved for more than 40 years. The many advantages of cryopreservation far outweigh the required investment in equipment and reagents. These advantages include:
- Generation of safety stocks to ensure against loss of the culture from equipment failures or contamination by microorganisms or other cell lines.
- Elimination of the time, energy, and materials required to maintain cultures not in immediate use.
- Preservation of cells with finite population doublings (that will ultimately senesce).
- Insurance against phenotypic drift in the culture due to genetic instability and/or selective pressure.
- Creating a standard reagent to be used for a series of experiments.
Overview
As the cell suspension is cooled below the freezing point, ice crystals form and the concentration of the solutes in the suspension increases. Intracellular ice can be minimized if water within the cell is allowed to escape by osmosis during the cooling process. A slow cooling rate, generally –1°C per minute, facilitates this process. However, as the cells lose water, they shrink in size and will quickly lose viability if they go beyond a minimum volume. The addition of cryoprotectant agents such as glycerol or dimethylsulfoxide (DMSO) will mitigate these effects.
The standard procedure for cryopreservation is to freeze cells slowly until they reach a temperature below –70°C in medium that includes a cryoprotectant. Vials are transferred to a liquid-nitrogen freezer to maintain them at temperatures below –130°C.
The recovery of cryopreserved cells is straightforward: Cells are thawed rapidly in a water bath at 37°C, removed from the freeze-medium by gentle centrifugation and/or diluted with growth medium, and seeded in a culture vessel in complete growth medium.
There are numerous factors which affect the viability of recovered cells. Modify the procedure for each cell line to attain optimal cell viability upon recovery. Some of the critical parameters for optimization include the composition of the freeze medium, the growth phase of the culture, the stage of the cell in the cell cycle, and the number and concentration of cells within the freezing solution.
ATCC provides information on cryopreservation for all cell lines on the Product Sheet. Most ATCC cell lines are frozen with a cryopreservation medium consisting of 5% DMSO and complete growth medium.
Freeze medium
Glycerol and DMSO at 5% to 10% are the most common cryoprotectant agents. While DMSO can be toxic to cells, it penetrates them much faster than glycerol and yields more reproducible results. Unfortunately, DMSO can cause some cells to differentiate (eg, HL-60 promyeloblast cells) and may be too toxic for some cells (eg, HBE4-E6/E7 lung epithelial cells). Glycerol should be used in these instances. Glycerol can be sterilized by autoclaving whereas DMSO must be sterilized by filtration. Care should be used when handling any DMSO solution as it will rapidly penetrate intact skin and may carry toxic contaminants along with it.
Use only reagent-grade (or better, such as cell culture-grade) DMSO or glycerol. Store both in aliquots protected from light. ATCC offers DMSO (ATCC 4-X) that has been thoroughly tested for cell culture use.
For cells grown in serum-free medium, adding 50% conditioned medium (serum-free medium in which the cells were grown for 24 hours) to both the cell freezing and the recovery medium may improve recovery and survival. The addition of 10% to 20% cell culture-grade bovine serum albumin to serum-free freezing medium may also increase postfreeze survival.
Serum-free freezing media have also been developed. ATCC Serum-Free Cell Freezing Medium (ATCC 30-2600) can be used for both cells cultured in serum-supplemented growth medium as well as cells grown under serum-free conditions. This proprietary formulation contains 10% DMSO and methylcellulose is suitable for the cryopreservation of adherent and suspension cell cultures. Cells cryopreserved using Serum-Free Freezing Medium show levels of viability and percent attachment (adherent cells) that are comparable to cells preserved in DMSO and FBS. (See: Figure 3)
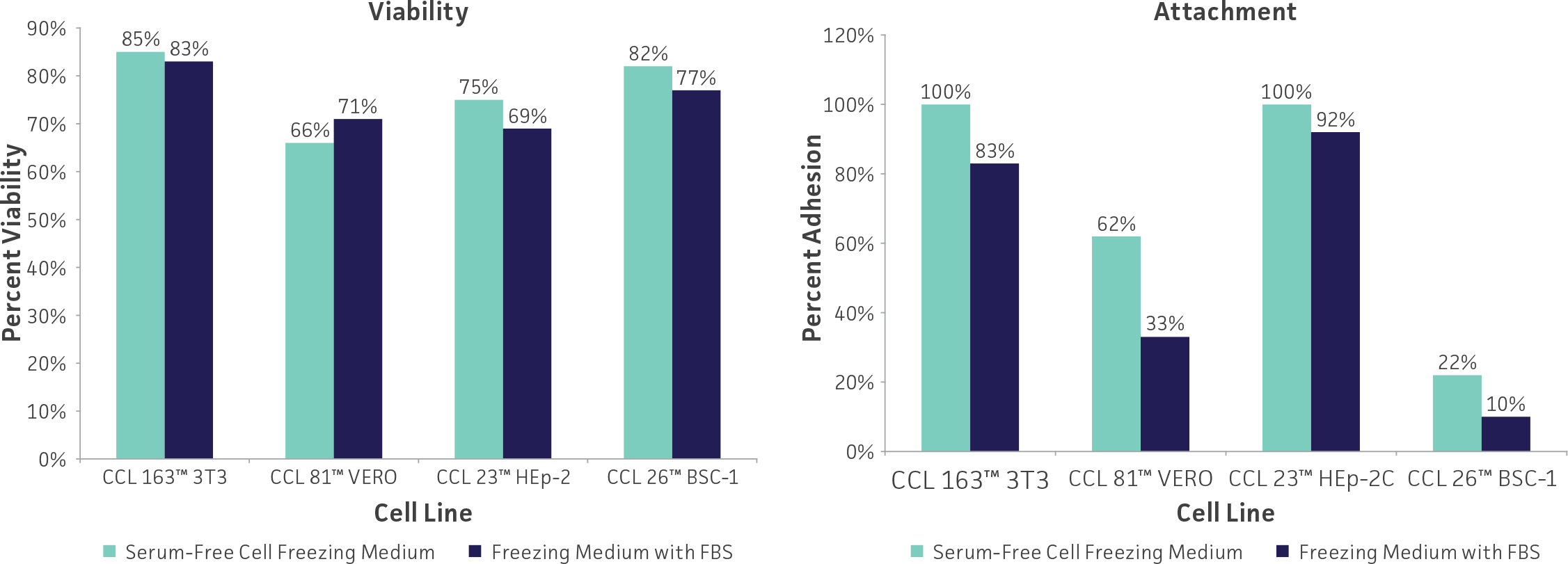
Figure 3: Cells cryopreserved using ATCC Serum-Free Cell Freezing Medium show levels of viability and percent attachment that are comparable to cells preserved in DMSO and FBS.
Other variations of freeze medium formulations include high (up to 90%) concentrations of serum which presumably supplies some cryoprotection as well as additional growth factors; use of a balanced salt solution designed for hypothermal conditions in place of medium designed for 37°C incubation; and the addition of apoptotic inhibitors which may prevent delayed onset cell death following recovery.21 Optimum formulations for individual cell lines need to be determined empirically.
Equipment
Cryopreservation vials
 There are two materials to choose from for cryopreservation vials: glass or plastic. Glass vials are more difficult to work with; they need to be sterilized before use, they do not come with labels (information is imprinted into the glass), they need to be sealed with a hot flame, and they can be difficult to open. However, they are preferred for long-term storage (many years) of valuable cultures and are considered fail-safe once properly sealed. ATCC uses glass vials for the storage of seed stocks which are placed in the lower level of the liquid nitrogen tank. Plastic vials are used for the storage of distribution stocks.
There are two materials to choose from for cryopreservation vials: glass or plastic. Glass vials are more difficult to work with; they need to be sterilized before use, they do not come with labels (information is imprinted into the glass), they need to be sealed with a hot flame, and they can be difficult to open. However, they are preferred for long-term storage (many years) of valuable cultures and are considered fail-safe once properly sealed. ATCC uses glass vials for the storage of seed stocks which are placed in the lower level of the liquid nitrogen tank. Plastic vials are used for the storage of distribution stocks.
Plastic vials come in two varieties: those with an internal thread and silicone gasket and those with an external thread. The internal-thread version was the first commercially available, but has some disadvantages over the external-thread version. For example, while the silicone gasket provides an excellent seal, it needs to be tightened just right; too tight or too loose and the vial will leak.
Controlled-rate freezing chambers
There are several means to achieve a cooling rate of –1°C per minute. The best is with a computer controlled, programmable electronic freezing unit (such as CryoMed Freeze) which rigorously maintains this rate of cooling. This is the method used exclusively at ATCC. Such equipment is relatively expensive and absolutely necessary for only the most sensitive cells.
A less costly approach is to place the cryopreservation vials into an insulated chamber and cool for 24 hours in a mechanical freezer at –70°C or lower. There are several commercially available freezing chambers which achieve a cooling rate very close to the ideal –1°C per minute (Mr. Frosty, Nalgene 5100-0001; or StrataCooler, Agilent Technologies 401349). Alternately, the vials can be placed into a polystyrene box with 15-mm (3/4 inch) thick walls and 1L capacity packed with paper, cotton wool, or foam peanuts for insulation.
Liquid nitrogen freezer storage
The ultra-low temperatures (below –130°C) required for long-term storage can be maintained by specialized electric freezers or more commonly by liquid nitrogen freezers. There are two basic types of liquid nitrogen storage systems: immersing vials in the liquid and holding vials in the vapor phase above the liquid. The liquid-phase system holds more nitrogen and thus requires less maintenance. However, there is always a chance that some liquid will enter improperly sealed vials which may explode when retrieved. For this reason ATCC strongly recommends storage in vapor-phase systems.
Vapor-phase systems create a vertical temperature gradient within the container. The temperature in the liquid nitrogen at the bottom will be –196°C, whereas the temperature at the top will vary depending upon the amount of liquid nitrogen at the bottom as well as the amount of time the container is opened. To ensure safe storage of cells, be sure to keep enough liquid nitrogen in the container so that the temperature at the top is –130°C or colder. All storage systems should be equipped with temperature alarms.
Cryopreservation procedure
The procedure below will work for most cell cultures and should be modified as needed. Freeze medium formulations for all ATCC cell lines are provided on the Product Sheet. Harvest cells in exponential growth.
- Check your cell culture for contamination from bacteria, fungi, mycoplasma, and viruses (see Contamination) immediately before cryopreservation. In most cases, the results of the contamination screen will be available some time after the cultures are cryopreserved (10 to 14 days). If contamination is confirmed, then destroy the frozen material.
- Prepare a freeze medium consisting of complete growth medium and 5% DMSO (ATCC 4-X). Do not add undiluted DMSO to a cell suspension as dissolution of DMSO in aqueous solutions gives off heat.
- Collect cells by gentle centrifugation (10 minutes at 125 × g) and resuspend them in the freeze medium at a concentration of 1 × 106 to 5 × 106 viable cells/mL. Continue to maintain the cells in culture until the viability of the recovered cells is confirmed (see Step 9).
- Label the appropriate number of vials with the name of the cell line and the date. Then add 1 to 1.8 mL of the cell suspension to each of the vials (depending upon the volume of the vial) and seal.
- Allow cells to equilibrate in the freeze medium at room temperature for a minimum of 15 minutes but no longer than 40. This time is usually taken up in dispensing aliquots of the cell suspension into the vials. After 40 minutes, cell viability may decline due to the DMSO.
- Place the vials into a pre-cooled (4°C), controlled-rate freeze chamber and place the chamber in a mechanical freezer at –70°C (or colder) for at least 24 hours. Alternately, use a pre-cooled (4°C) programmable freezer unit set to cool the vials at –1°C per minute until a temperature below –40°C is achieved and then set to abruptly drop to –130°C.
- Quickly transfer the vials to a liquid nitrogen or –130°C freezer. Frozen material will warm up at a rate of 10°C per minute and cells will deteriorate rapidly if warmed above –50°C.
- Record the location and details of the freeze.
- After 24 hours at –130°C, remove one vial, restore the cells in culture medium, and determine their viability and sterility.
Recovery of cryopreserved cells
The cell solution in the frozen vial needs to be warmed as rapidly as possible and then immediately combined with complete culture medium and seeded into an appropriate flask. While cells grown in monolayers can be recovered from cryopreservation in multiwell plates, the results are not as consistent as with flasks.
Some cell lines, such as hybridoma cultures, take several days before they fully recover from cryopreservation. Some hybridomas show low viability on the first day in culture and will generate cellular debris. Viability for most cells declines and reaches a nadir at 24 hours post-thaw. Most, if not all, of this decline appears to be due to apoptosis (as opposed to necrosis) induced by the stress of the cryopreservation process.22 After this time point, cells begin to recover and enter exponential growth.
- Prepare a culture vessel (T-75 flask) so that it contains at least 10 mL of the appropriate culture medium equilibrated for temperature and pH.
- Remove the vial from the liquid nitrogen freezer and thaw by gentle agitation in a 37°C water bath (or a bath set at the normal growth temperature for that cell line). Thaw rapidly until ice crystals have melted (approximately 2 minutes).
- Remove the vial from the water bath and decontaminate it by dipping in or spraying with 70% ethanol. Follow strict aseptic conditions in a laminar flow tissue culture hood for all further manipulations.
- Unscrew the top of the vial and transfer the contents to a sterile centrifuge tube containing 9 mL complete growth medium. Remove the cryoprotectant agent by gentle centrifugation (10 minutes at 125 × g). Discard the supernatant, taking care not to disturb the soft pellet, and resuspend the cells in 1 mL or 2 mL of complete growth medium. Pipette gently to loosen the pellet and break apart clumps. (If the cells normally grow as clusters, avoid over-pipetting during resuspension.) Transfer the cell suspension into the medium in the culture vessel and mix thoroughly.
- Examine the cultures after 24 hours and subculture as needed.

Contamination and biosafety
Contamination of cells in culture can arise from many sources including other cell lines, reagents, supplies such as pipettes and culture vessels, equipment such as tissue culture hoods and incubators, and laboratory personnel. While the potential for contamination is constant, the risk can be reduced or eliminated by proper precautions: using only reagents of known quality and sterility, quarantining new cell lines until they are tested to be free from contamination, performing routine maintenance and cleaning of all equipment, and properly training cell culture personnel.
Check for microbial contamination
When most bacterial contamination occurs, it usually occurs within a few days and is typically obvious to the naked eye. Distinct changes to the medium such as turbidity, presence of particles visible in suspension, and a rapid decline in pH (yellow color, indicating acidity) are all indicators of bacterial contamination. Fastidious bacteria species that grow very slowly can be difficult to detect.
Fungal contaminants may or may not cause a change in the pH of the medium and can be distinguished from bacteria by checking for the presence of filamentous structures in the suspension. Yeast cells are larger than bacteria, but may not appreciably change the pH of the medium, and will appear as separate round or ovoid particles. Microbacterial media which can be used to test for bacterial and fungal contamination include blood agar, thioglycollate broth, tryptic soy broth, BHI broth, Sabouraud broth, YM broth, and nutrient broth with 2% yeast extract.23 However, some microbial contamination is not apparent. For example, the use of antibiotics can suppress bacterial growth and thus mask contamination. Some viral infections do not alter the morphology of the cells, and detection of mycoplasma contamination requires specific assays.
Mycoplasma contamination
Cell lines are screened for mycoplasma contamination by direct (agarose and broth culture) and indirect (Hoechst) methods.24,25 For example, the fluorochrome Hoechst DNA stain will bind to the DNA of mycoplasma and the organisms can be detected easily when examined using a microscope equipped with appropriate fluorescence optics. The direct culture method requiring both broth and agar will permit isolation of cultivable strains as apparent by appearance of characteristic mycoplasma colonies on the agar medium.
Both direct and indirect methods for detection of mycoplasma are used at ATCC several times while a cell line is expanded for the preparation of the token, seed and distribution stocks.
Most cell culture laboratories have incorporated PCR-based mycoplasma testing, using kits such as ATCC’s Universal Mycoplasma Detection Kit (ATCC 30-1012K) into their routine cell culture operations.
Cell cultures can be submitted to the ATCC Mycoplasma Testing Service. See the ATCC Services section of the website for details.
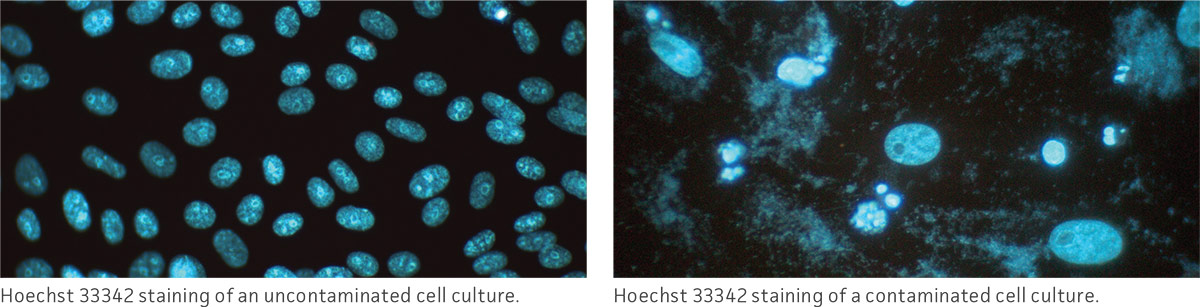
Treating for microbial contamination
Eliminating contamination from a cell line is time consuming and does not always work. Discarding the culture and starting over is preferred. However, if the cells are unique and irreplaceable, one should first identify the contaminant and select a suitable antibiotic for treatment. It is best to test the contaminating microbe for its antibiotic sensitivity prior to treatment; this allows for a shorter treatment time and limits exposure of the cell line to potentially damaging reagents.
The cells are cultured for 1 to 2 weeks in the presence of the antibiotic, and then cultured without antibiotic for 1 to 2 months. At this point, the line should be retested with a very sensitive test method to make sure that the culture is clean. Periodic retesting should be employed to make sure that the contaminant does not reappear. Since antibiotics may be toxic to cells, a selected population that no longer exhibits qualities of the parental line may result. It may be necessary to examine the cured culture to determine if it is sufficiently similar to the original line.
Cellular cross-contamination
Cross-contamination of one cell line with another can sometimes lead to the replacement of the original cell with the contaminant, particularly when the contaminant grows faster than the original line. HeLa cells are perhaps the most famous example of a cross-contaminating cell line overtaking and then masquerading as the original.
In the 1950s and 1960s, many continuous lines were unknowingly cross-contaminated with other cell lines including HeLa cells. In the 1970s and 1980s, as many as one in three cell lines deposited in cell repositories were imposters.26 This cross-contamination was only uncovered with the development of suitable genetic markers beginning in 1967.27 Indeed, several “unique” cell lines in ATCC’s collection turned out to be HeLa cells upon further study. Despite the confirmation of their HeLa cell origin, cytogenetic analysis suggests that there are differences among these HeLa-derived cell lines. Several of them possess unique properties. However, these cell lines should not be used as functional models of their claimed tissues of origin.
More recently, ATCC and other cell repositories have used DNA polymorphisms in addition to enzyme polymorphisms, HLA typing, and karyotyping to confirm the identity of their cell lines. One of the most reliable methods to study DNA polymorphisms is the profiling of short tandem repeats (STR) by PCR amplification followed by capillary electrophoresis.28 STR profiles for all ATCC human cell lines are available on the website in the catalog descriptions. (See: Figure 4)
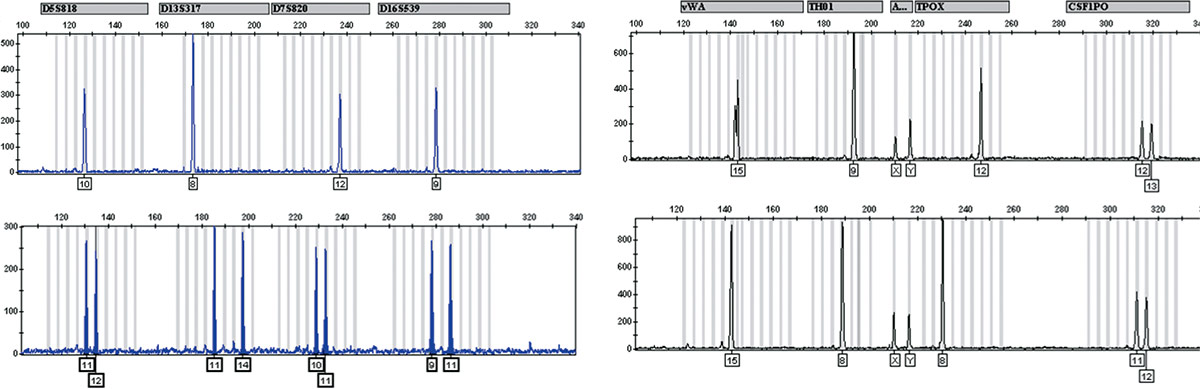
Figure 4: STR profiles for two unrelated human cell lines. Top: KU812E (ATCC CRL-2100). Bottom: MRC-5 (ATCC CCL-171). Amplicons were generated using Promega’s PowerPlex platform, separated by capillary electrophoresis and analyzed using GeneMapper software from Life Technologies.
Test cell cultures on a regular basis to ensure the absence of contamination from both microorganisms as well as from other cell lines. If contamination is found, discard the culture and start fresh with a new stock.
Biosafety
The need for precautions when experimenting with cells in culture depends upon the source and nature of the biological material, the experimental procedure, and the laboratory/containment conditions. Since every situation is different, the risks need to be identified and appropriate precautions need to be taken before any work begins.
More information on risk assessment and precautions can be found in the Center for Disease Control (CDC) publication Biosafety in Microbiological and Biomedical Laboratories, (BMBL) 6th Edition.29 The text of this publication is available in its entirety online (https://www.cdc.gov/labs/bmbl/index.html). Information on agent risk assessment and a description of the four biosafety levels can be found in this publication.
ATCC assigns a biosafety level (BSL) to each cell line for purposes of packaging for safe shipment. When a cell line is known to contain an etiologic agent, ATCC classification is at least comparable to the BSL assigned to the agent by the CDC and in some cases the ATCC designation is more restrictive. ATCC follows federal biosafety guidelines and takes several factors into consideration when assessing potential hazard.
Biosafety Level 1
- Cell lines with animal origin not included under Biosafety Level 2
Biosafety Level 2
- Cell lines that harbor mycoplasma or any other BSL 2 agent (See: NOTE 7)
- Cell lines exposed to or transformed by a primate oncogenic virus
- Primate cell lines that contain viruses
- Cell lines carrying a part of certain viral genomes, even if whole virus is not released from the cell30
As the recipient of a cell line, take into account not only the nature of the material but also the manipulations employed during its handling when assessing the potential laboratory risk. For example, procedures involving large volumes of cell lines that contain HIV or that include manipulation of HIV in high concentration should be conducted under BSL 3 conditions.29
NOTE 7
It is not possible to screen cell lines for the presence of every agent. For added precaution, ATCC handles all cell lines under BSL 2 practices, even those classified as BSL 1. It is prudent to treat all mammalian cell lines as potentially hazardous.

ATCC media, sera, and reagents
Table 1: ATCC media product list
| ATCC number | Product name | Volume |
|---|---|---|
| 30-2002 | Dulbecco’s Modified Eagle’s Medium (DMEM) | 500 mL |
| 30-2006 | DMEM:F12 Medium (1:1 Ratio) | 500 mL |
| 30-2003 | Eagle’s Minimum Essential Medium (EMEM) | 500 mL |
| 30-2004 | F12K Medium | 500 mL |
| 46-X | Hybri-Care Medium, Powder | Prepares 1L |
| 30-2005 | Iscove’s Modified Dulbecco’s Medium (IMDM) | 500 mL |
| 30-2008 | Leibovitz’s L-15 Medium | 500 mL |
| 30-2007 | McCoy’s 5A Medium, Modified | 500 mL |
| 30-2001 | RPMI-1640 Medium | 500 mL |
*See complete media formulations.
Table 2: Animal sera product list
| ATCC number | Product name | Volume |
|---|---|---|
| 30-2020 | Fetal Bovine Serum | 500 mL |
| 30-2021 | Fetal Bovine Serum | 100 mL |
| SCRR-30-2020 | Fetal Bovine Serum, ES Qualified* | 500 mL |
| 30-2030 | Calf Bovine Serum | 500 mL |
| 30-2031 | Calf Bovine Serum | 100 mL |
*Qualified for mouse and human embryonic stem cells.
Table 3: Supplements and antibiotics product list
| ATCC number | Product name | Volume |
|---|---|---|
| 30-2214 | L-Glutamine Solution | 100 mL |
| PCS-999-002 | Penicillin-Streptomycin-Amphotericin B Solution | 1.0 mL |
| 30-2300 | Penicillin-Streptomycin Solution | 100 mL |
Table 4: Buffers, stains and dissociation reagents product list
| ATCC number | Product name | Volume |
|---|---|---|
| 30-2103 | Non-Enzymatic Cell Dissociation Solution | 100 mL |
| 30-2104 | Soybean Trypsin Inhibitor | 20 mL |
| 30-2101 | 0.25% Trypsin/0.53 mM EDTA Solution | 100 mL |
| PCS-999-003 | Trypsin-EDTA for Primary Cells* | 100 mL |
| PCS-999-004 | Trypsin Neutralizing Solution | 100 mL |
| 30-2200 | Dulbecco’s Phosphate Buffered Saline (DPBS)* | 500 mL |
| 30-2213 | Hank’s Balanced Salt Solution (HBSS)* | 500 mL |
| PCS-999-001 | Phenol Red | 1.0 mL |
Table 5: Cryopreservation reagents product list
| ATCC number | Product name | Volume |
|---|---|---|
| 4-X | Dimethylsulfoxide (DMSO) | 5 × 5 mL |
| 30-2600 | Serum-Free Cell Freezing Medium | 20 mL |
Table 6: Supplements and antibiotics product list
| ATCC number | Product name | Volume |
|---|---|---|
| PCS-999-027 | 0.1% Gelatin Solution | 100 mL |
Table 7: Cell proliferation assays and mycoplasma detection
| ATCC number | Product name |
|---|---|
| 30-1010K | ATCC MTT Cell Proliferation Assay |
| 30-1011K | XTT Cell Proliferation Assay Kit |
| 30-1012K | Universal Mycoplasma Detection Kit |
Download a PDF of our Animal Cell Culture Guide
Download Now
Glossary
The following glossary was originally published by the Tissue Culture Association Terminology Committee in 1990.31
Anchorage-dependent cells or cultures. Cells, or cultures derived from them, which will grow, survive, or maintain function only when attached to a surface such as glass or plastic. The use of this term does not imply that the cells are normal or that they are not neoplastically transformed.
Aneuploid. The situation in which the nucleus of a cell does not contain an exact multiple of the haploid number of chromosomes, one or more chromosomes being present in greater or lesser number than the rest. The chromosomes may or may not show rearrangements.
Aseptic technique. Procedures used to prevent the introduction of fungi, bacteria, viruses, mycoplasma, or other microorganisms in cell, tissue, and organ cultures. Although these procedures are used to prevent microbial contamination of cultures, they also prevent cross-contamination of cell cultures as well.
Attachment efficiency. The percentage of cells plated (seeded, inoculated) which attach to the surface of the culture vessel within a specified period of time. The conditions under which such a determination is made should always be stated.
Autocrine cell. In animals, a cell which produces hormones, growth factors, or other signaling substances for which it also expresses the corresponding receptors. (See also endocrine and paracrine.)
Cell culture. Term used to denote the maintenance or cultivation of cells in vitro including the culture of single cells. In cell cultures, the cells are no longer organized into tissues.
Cell generation time. The interval between consecutive divisions of a cell. This interval can best be determined, at present, with the aid of cinephotomicrography. This term is not synonymous with population doubling time.
Cell hybridization. The fusion of two or more dissimilar cells leading to the formation of a synkaryon.
Cell line. A cell line arises from a primary culture at the time of the first successful subculture. The term implies that cultures from it consist of lineages of cells originally present in the primary culture. The terms finite or continuous are used as prefixes if the status of the culture is known. If not, the term line will suffice. The term continuous line replaces the term established line. In any published description of a culture, one must make every attempt to publish the characterization or history of the culture. If such has already been published, a reference to the original publication must be made. In obtaining a culture from another laboratory, the proper designation of the culture, as originally named and described, must be maintained and any deviations in cultivation from the original must be reported in any publication.
Cell strain. A cell strain is derived either from a primary culture or a cell line by the selection or cloning of cells having specific properties or markers. In describing a cell strain, its specific features must be defined. The terms finite or continuous are to be used as prefixes if the status of the culture is known. If not, the term strain will suffice. In any published description of a cell strain, one must make every attempt to publish the characterization or history of the strain. If such has already been published, a reference to the original publication must be made. In obtaining a culture from another laboratory, the proper designation of the culture, as originally named and described, must be maintained and any deviations in cultivation from the original must be reported in any publication.
Chemically defined medium. A nutritive solution for culturing cells in which each component is specifiable and, ideally, is of known chemical structure.
Clone. In animal cell culture terminology, a population of cells derived from a single cell by mitoses. A clone is not necessarily homogeneous and therefore the terms clone and cloned do not indicate homogeneity in a cell population, genetic or otherwise.
Cloning efficiency. The percentage of cells plated (seeded, inoculated) that form a clone. One must be certain that the colonies formed arose from single cells in order to properly use this term. (See colony forming efficiency.)
Colony forming efficiency. The percentage of cells plated (seeded, inoculated) that form a colony.
Contact inhibition of locomotion. A phenomenon characterizing certain cells in which two cells meet, locomotory activity diminishes and the forward motion of one cell over the surface of the other is stopped.
Continuous cell culture. A culture which is apparently capable of an unlimited number of population doublings, often referred to as an immortal cell culture. Such cells may or may not express the characteristics of in vitro neoplastic or malignant transformation. (See also immortalization.)
Crisis. A stage of the in vitro transformation of cells. It is characterized by reduced proliferation of the culture, abnormal mitotic figures, detachment of cells from the culture substrate, and the formation of multinucleated or giant cells. During this massive cultural degeneration, a small number of colonies usually, but not always, survives and gives rise to a culture with an apparent unlimited in vitro lifespan. This process was first described in human cells following infection with an oncogenic virus (SV40). (See also cell line, in vitro transformation, and in vitro senescence.)
Cryopreservation. Ultra-low temperature storage of cells, tissues, embryos, or seeds. This storage is usually carried out using temperatures below –100°C.
Density-dependent inhibition of growth. Mitotic inhibition correlated with increased cell density.
Differentiated. Cells in culture that maintain all or much of the specialized structure and function typical of the cell type in vivo.
Diploid. The state of the cell in which all chromosomes, except sex chromosomes, are two in number and are structurally identical with those of the species from which the culture was derived.
Electroporation. Creation by means of an electrical current of transient pores in the plasmalemma usually for the purpose of introducing exogenous material, especially DNA, from the medium.
Embryo culture. In vitro development or maintenance of isolated mature or immature embryos.
Embryogenesis. The process of embryo initiation and development.
Endocrine cell. In animals, a cell which produces hormones, growth factors or other signaling substances for which the target cells, expressing the corresponding receptors, are located at a distance. (See also autocrine or paracrine.)
Epithelial-like. Resembling or characteristic of, or having the form or appearance of, epithelial cells. In order to define a cell as an epithelial cell, it must possess characteristics typical of epithelial cells. Often one can be certain of the histologic origin and/or function of the cells placed into culture and, under these conditions, one can be reasonably confident in designating the cells as epithelial. The individual reporting on such cells should use as many parameters as possible in assigning this term to a culture. Until a rigorous definition is possible, it is more correct to use the term epithelial-like.
Euploid. The situation in which the nucleus of a cell contains exact multiples of the haploid number of chromosomes.
Feeder layer. A layer of cells (usually irradiated or mitomycin-C treated) that are nondividing but metabolically active, upon which a fastidious cell type is cultured.
Finite cell culture. A culture which is capable of only a limited number of population doublings after which the culture ceases proliferation. (See in vitro senescence.)
Heterokaryon. A cell possessing two or more genetically different nuclei in a common cytoplasm, usually derived as a result of cell-to-cell fusion.
Heteroploid. A culture whose cells contain chromosome number other than the diploid number. This is a term used only to describe a culture and is not used to describe individual cells. Thus, a heteroploid culture would be one which contains aneuploid cells.
Histiotypic. The in vitro resemblance of cells in culture to a tissue in form, function, or both. For example, a suspension of fibroblast-like cells may secrete a glycosaminoglycan-collagen matrix and the result is a structure resembling fibrous connective tissue, which is, therefore, histiotypic. This term is not meant to be used along with culture. Thus, a tissue culture system demonstrating form and function typical of the cells in vivo would be said to be histiotypic.
Homokaryon. A cell possessing two or more genetically identical nuclei in a common cytoplasm, derived as a result of cell-to-cell fusion.
Hybridoma. The cell which results from the fusion of an antibody-producing tumor cell (myeloma) and an antigenically stimulated normal plasma cell. Such cells are constructed because they produce a single antibody directed against the antigen epitope which stimulated the plasma cell. This antibody is referred to as a monoclonal antibody.
Immortalization. The attainment by a cell culture, whether by perturbation or intrinsically, of the attributes of a continuous cell line. An immortalized cell is not necessarily one which is neoplastically or malignantly transformed.
In vitro senescence. The inability of a vertebrate cell culture to grow beyond a finite number of population doublings. Neither invertebrate nor plant cell cultures exhibit this property.
In vitro transformation. A heritable change occurring in cells in culture, either intrinsically or from treatment with chemical carcinogens, oncongenic viruses, irradiation, transfection with oncogenes, etc., which leads to the acquisition of altered morphological, antigenic, neoplastic, proliferative, or other properties. This expression is distinguished from in vitro neoplastic transformation in that the alterations occurring in the cell population may not always include the ability of the cells to produce tumors in appropriate hosts. The type of transformation should always be specified in any description.
Organ culture. The maintenance or growth of organ primordia or the whole or parts of an organ in vitro in a way that may allow differentiation and preservation of the architecture and/or function.
Paracrine. In animals, a cell which produces hormones, growth factors or other signaling substances for which the target cells, expressing the corresponding receptors, are located in its vicinity, or in a group adjacent to it. (See also autocrine and endocrine.)
Passage. The transfer or transplantation of cells, with or without dilution, from one culture vessel to another. It is understood that any time cells are transferred from one vessel to another, a certain portion of the cells may be lost, and therefore dilution of cells, whether deliberate or not, may occur. This term is synonymous with subculture.
Passage number. The number of times the cells in the culture have been subcultured or passaged. In descriptions of this process, the ratio or dilution of the cells should be stated so that the relative cultural “age” can be ascertained.
Plating efficiency. This term originally encompassed the terms attachment efficiency, cloning efficiency, and colony forming efficiency; it is now better to use one or more of them in its place because plating is not sufficiently descriptive. (See attachment efficiency, cloning efficiency, and colony forming efficiency.)
Population density. The number of cells per unit area or volume of a culture vessel, or the number of cells per unit volume of medium in a suspension culture.
Population doubling level. The total number of population doublings of a cell line or strain since its initiation in vitro. This term is synonymous with cell generation time.
Population doubling time. The interval, calculated during the logarithmic phase of growth in which cells double in number; for example, 1.0 x 106 cells increase to 2.0 x 106 cells. This term is not synonymous with cell generation time.
Primary culture. A culture started from cells, tissues, or organs taken directly from organisms. A primary culture may be regarded as such until it is successfully subcultured for the first time. It then becomes a cell line.
Pseudodiploid. The condition in which the number of chromosomes in a cell is diploid but, as a result of chromosomal rearrangements, the karyotype is abnormal and linkage relationships may be disrupted.
Saturation density. The maximum cell number attainable, under specified culture conditions, in a culture vessel. This term is usually expressed as the number of cells per square centimeter in a monolayer culture or the number of cells per cubic centimeter in a suspension culture.
Suspension culture. A type of culture which will grow and can be maintained without attaching to a surface, such as glass or plastic.
Transfection. The transfer, for the purpose of genomic integration, of foreign DNA into cells in culture. The traditional microbiological usage of this term implied that the DNA being transferred was derived from a virus. The definition as stated here describes the general transfer of DNA irrespective of its source.
Undifferentiated. With animal cells, this is the state wherein the cell in culture lacks the specialized structure and/or function of the cell type in vivo.

References
- Hayflick and Moorhead PS. Exp. Cell Res. 25: 585-621, 1961.
- Rubin H. Mech. Aging Devel. 98: 1-35, 1997.
- Wright WE and Shay JW. Nat. Biotechnol. 20: 682-688, 2002.
- Capstick PB et al. Exp. Cell Res. 44: 119-128, 1966.
- Osato et al. J. Immunology 124: 533-540, 1980.
- McLimans WF. Chapter 5. In: Growth, Nutrition and Metabolism of Cells in Culture, Vol.1. Rothblat GH and Cristofalo VJ, eds. New York: Academic Press; 1972.
- Freshney RI. Chapter 8. In: Culture of Animal Cells, 5th ed. New York: Wiley-Liss; 2005.
- Shipman C. Proc. Soc. Exp. Biol. Med. 130: 305, 1969.
- People CA et al. In Vitro 18: 755, 1982.
- Spierenberg GT et al. Cancer Res. 44: 2253, 1984.
- Zigler JS et al. In Vitro 21: 282, 1985.
- Karmiol S. Development of serum free media. In: Master JRW, ed. Animal Cell Culture, 3rd ed. Oxford: Oxford University Press; 2000.
- Jacoby WB and Pasten IH, eds. Chapter 7. In: Methods in Enzymology: Cell Culture, Vol. 58. New York: Academic Press; 1979.
- Bolin SR et al. J. Virol. Methods 48: 211, 1994.
- Hyclone, Inc. Art to Science 15(1): 1-6, 1996.
- Rudnicki MA and McBurney MW. Teratocarcinomas and Embryonic Stem Cells - A Practical Approach. Oxford: IRL Press Ltd.; 1987: p. 75.
- Weiss SA et al. Meth. Mol. Biol. 39: 65, 1995.
- Gabridge MG. Vessels for Cell and Tissue Culture. In: Setting Up and Maintenance of Tissue and Cell Cultures. Shannon: Elsevier Scientific Publishers; 1985: pp. 1-19.
- Balin A.K et al. Atmospheric Stability in Cell Culture Vessels. In Vitro 12: 687-692, 1977.
- Gey GO. Am. J. Cancer 17: 752-756, 1933.
- Baust JM et al. Cell Preservation Technology 1: 17-31, 2002.
- Baust JM et al. Cell Preservation Technology 1: 63-80, 2002.
- Quality Control Methods for Cell Lines, 2nd edition. Rockville, MD: ATCC; 1992.
- Freshney RI. Culture of Animal Cells, 5th ed. New York: Wiley-Liss; 2005.
- Lincoln CK and Gabridge MG. Cell culture contamination: Sources, consequences, prevention and elimination. In: Mather JP and Barnes D, eds. Animal Cell Culture Methods. San Diego: Academic Press; 1998: p. 49.
- O’Brien SJ. Science 98: 7656-7658, 2001.
- Gartler SM. NCI Monograph 26: 167-195, 1967.
- Master JR. et al. Proc. Natl. Acad. Sci. USA 98: 8012-8017, 2001.
- U.S. Department of Health and Human Services, Centers for Disease Control and Prevention. Biosafety in Microbiological and Biomedical Laboratories, 6th ed. HHS Publication No. (CDC) 300859. Washington DC: U.S. Government Printing Office; 2020. Available online at https://www.cdc.gov/labs/pdf/SF__19_308133-A_BMBL6_00-BOOK-WEB-final-3.pdf.
- Weiss RA. NCI Monograph 48: 183-189, 1978.
- Schaeffer WI. In Vitro Cell. Dev. Biol. 26: 97-101, 1990.