Use our detailed guide to access tips and techniques for the growth, propagation, preservation, and safe handling of protists.
Table of Contents
Getting Started with an ATCC Protist Strain
Protist Growth and Propagation
Growth Media
Preservation
Biosafety and Disposal
Protist Authentication
Protist Applications
Glossary
References
Download a PDF of our protistology culture guide
Get the guideGetting started with an ATCC protist strain
ATCC protist strains are shipped frozen on dry ice in plastic cryopreservation vials, as freeze-dried cultures in glass ampoules or serum vials, or as live cultures in test tubes. Upon receipt of a frozen culture, immediately revive cells by thawing and transferring to an appropriate growth medium. If this is not possible, store frozen vials in liquid nitrogen vapor (below -130°C). Alternatively, the frozen material can be stored between -70°C and -80°C for short periods (1 to 5 days); however, viability will decline at temperatures above -130°C. Upon receipt of a freeze-dried culture, rehydrate cultures using the appropriate medium and incubation conditions specified on the product sheet. Freeze-dried cultures can be stored safely at 4°C or colder. Live cultures should be immediately handled according to the recommended protocols for that specific organism. Under no circumstances should live cultures be stored at refrigerated or frozen temperatures as this may result in the death of the culture.
Product sheet
ATCC protist strains typically have detailed information on the processing and expansion of materials, recommended media, as well as recommended growth and propagation conditions. The complete information for a product and the product sheet can be found on the product detail page for that specific material. Please note that ATCC will only warrant cultures that are handled according to the instructions provided on the ATCC product sheet. Product sheets, as well as additional information, can be found on the ATCC website or can be requested from the ATCC Product Experience team.
Preparation of medium
In advance, prepare the appropriate medium and additional supplements necessary for algal or protozoan revival and growth. Additionally, ensure that your incubator is set to maintain the optimal growth conditions of the strain and that any required light is available. Information for the formulation and preparation of media is available on the ATCC website.
Opening glass ampoules

Overview
All cultures should be considered potentially hazardous and should be opened by individuals trained in microbiological techniques working in facilities with containment requirements appropriate for the biosafety level of the cultures. ATCC recommends that the handling or opening of glass ampoules be performed in a biological safety cabinet. If this is not possible, wear protective clothing, gloves, a face shield or safety goggles, and hold the vial away from your body. Ensure that all empty vials are sterilized before disposal.
- Opening double-vial preparations
- Heat the tip of the vial in a flame.
- Add a few drops of water to the hot tip to crack the glass.
- Strike the end of the vial with a file or pencil to remove the tip.
- Remove the insulation and inner vial with sterile forceps. Gently raise the cotton plug.
- Opening single-vial preparations
- Recover the culture suspension from the glass ampoule by scoring the neck of the ampoule with a small, sterile file.
- Disinfect the outside of the ampoule with freshly prepared 70% ethanol or dip it into a beaker of freshly prepared 70% ethanol.
- Wrap the ampoule within several folds of a sterile towel or gauze to dry residual ethanol.
- Hold the vial upright and snap the vial open within a laminar flow hood. Ensure that the gauze does not become too wet with ethanol, or alcohol could be sucked into the culture when the vacuum is broken.

Initiating frozen cultures
- Prepare a sterile culture tube or flask that contains the recommended growth medium described in the product sheet. Ensure that the medium contains all necessary supplements or components and is equilibrated for temperature and pH.
- Thaw the sample vial in a water bath that is set to 35°C. Thawing will be rapid; approximately 2 minutes or until all ice crystals have melted. Protists should not be thawed at room temperature or in the refrigerator.
- Remove the vial from the water bath and decontaminate the outer surface using freshly prepared 70% ethanol. Follow strict aseptic conditions in a laminar flow hood for all further manipulations.
- Unscrew the top of the vial and transfer the entire contents to a culture tube or flask containing the appropriate growth medium.
- Incubate cultures under the appropriate temperature and atmospheric conditions as recommended on the product sheet.
- Monitor the cultures by microscopic examination following initiation. Once growth has stabilized, cultures can be maintained as described on the product sheet. Please note that the incubation period will vary between strains (See: NOTE 1).
Note 1:
Following recovery from a freeze-dried or frozen state, some protist strains may require an extended incubation period.
Initiating freeze-dried cultures
- Rehydrate freeze-dried strains by aseptically adding 0.5 mL of the recommended medium to the vial. For sensitive strains, rehydrate freeze-dried material using medium containing 12% sucrose.
- Aseptically transfer the entire contents of the vial to a sterile culture tube or flask containing the appropriate growth medium.
- Incubate the culture under the appropriate temperature and atmospheric conditions as recommended on the product sheet.
- Examine cultures microscopically following initiation. Once growth has stabilized, cultures can be maintained as described on the product sheet. Please note that the incubation period will vary between strains (See: NOTE 1).
Handling live protist cultures upon arrival
- Immediately place the culture under the recommended atmospheric conditions and temperature and allow culture to equilibrate for 1-3 hours or as recommended in the product sheet for the specific organism.
- Following equilibration, examine the culture to confirm its viability and healthy morphology, then handle as directed in the product sheet for the specific item.
- Examine cultures again after the recommended incubation period (See: NOTE 2). The incubation period will vary between strains and is listed on the product sheet. Follow the procedure for maintenance of the culture as directed in the product sheet for the specific item.
Note 2:
Some live cultures need to be incubated for a few hours upon receipt, then passaged as necessary. This will vary by taxon.

Protist growth and propagation
Properties of protists
Protists are a diverse group of eukaryotic organisms that includes the protozoa and algae. These organisms are of clinical relevance as many protozoan species are human pathogens, causing diseases such as malaria, giardiasis, and toxoplasmosis. Additionally, protists are of ecological importance as they are the predominant primary producers in most aquatic and soil environments and many species function as bacterial consumers.
In 2005 the taxonomic classification of protists was revised to recognize six clusters of eukaryotes representing the basic groupings similar to the traditional “kingdoms.” This taxonomic revision incorporated results obtained from ultrastructural studies and molecular phylogenetic studies to characterize protists into “supergroups.” The six supergroups were named Rhizaria, Opisthokonta, Archaeplastidia, Amoebozoa, Excavata, and Chromalveolata1 (Table 1). A more recent revision of eukaryote classification published in 2012 retained most of the super-groups proposed in 2005, although some have been assembled into still higher order groupings.2
Table 1. Protist supergroups*
| Supergroup | Characteristics | Examples** | Image |
|---|---|---|---|
| Amoebozoa | Amoeboid locomotion using blunt, oblong pseudopodia. Most species are unicellular and are common to aquatic and soil habitats.1 |
Tubulinea Flabellinea Stereomyxida Acanthamoebidae Entamoebida Mastigamoebidae Eumycetozoa |
ATCC 50924 Vannella cirrifera |
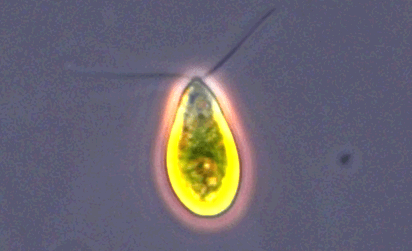
| Archaeplastida | Red and green algae, containing photosynthetic plastid with chlorophyll and pigments.1 | Glaucophyta Rhodophyceae Chloroplastida |
ATCC 30929 Dunaliella tertiolecta |
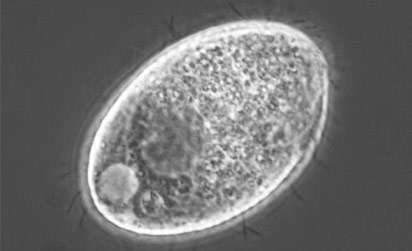
| Chromalveolata | Species contain a plastid obtained by secondary endosymbiosis with an ancestral archaeplastid.1 | Cryptophyceae Haptophyta Stramenopiles |
ATCC 205082 Tetrahymena ellioti |
| Excavata | Unicellular eukaryotes that include symbiotic, parasitic, and free-living forms. Many excavates lack classical mitochondria.1,3 | Fornicata Parabasalia Preaxostyla Jakobida Heterolobosea Euglenozoa |
ATCC 50286 Trepomonas agilis |
| Rhizaria | Amoeboid in form with filose, rediculose, or microtubule-supported pseudopodia.1,4 | Cercozoa Haplosporidia Foraminifera Radiolaria |
ATCC 50344 Cercomonas longicauda |
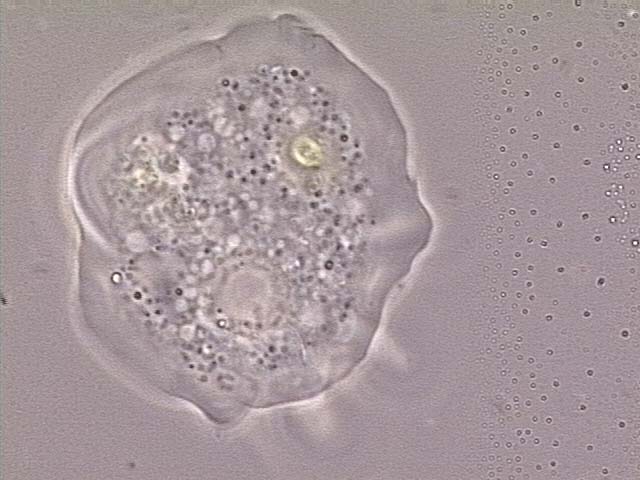
| Opisthokonta | Heterotrophic organisms having a single posterior cilium that is present in at least one life stage cycle.1,5 | Mesomycetoza Choanomonada Metazoa |
ATCC 50964 Monosiga gracilis |
*Classification is based on the 2005 revision of the classification of unicellular eukaryotes by the International Society of Protistologists.1
**Examples do not include a complete accounting of the full taxonomic diversity of the ATCC Protistology Collection but rather the most common protist groups housed by ATCC.
In addition to characterization by taxonomic classification, at ATCC we find it helpful to categorize groups of protists based on how they are handled (Table 2). This system of categorization allows us to generalize protocols and techniques for organisms with similar needs based on the culture system used (ie, whether the culture is xenic or axenic, whether liquid or solid media are used, the type of atmosphere required, etc.).
Table 2. Types of protist cultures
| Type of culture | Recommended handling | Examples* |
|---|---|---|
| Algae | Axenic culture grown in Erlenmeyer flasks or test tubes. Some strains can be cultured on plates or slants. | Chlorella, Chlamydomonas, Dunaliella, Haematococcus, Chlorogonium, Scenedesmus, Euglena |
| Free-living Amoebae | Aerobic xenic culture, exclusively grown on plates. | Xenic strains of Acanthamoeba, Naegleria, Hartmannella, Willaertia |
| Free-living Amoebae | Aerobic xenic cultures grown both in flasks and on plates. | Xenic strains of Vannella, Flabellula, Korotnevella, Paramoeba, Neoparamoeba, Platyamoeba, Vexillifera |
| Free-living Amoebae | Monoxenic or axenic, bacteria-free culture, exclusively grown in flasks. Few species are propagated in cell culture within flasks at a 5% CO2 atmosphere. | Monoxenic or axenic, bacteria-free strains of Acanthamoeba, Naegleria, Hartmannella, Willaertia, Vahlkampfia, Balamuthia |
| Ciliates | Xenic or axenic culture. Strains can be grown in both flasks and in tubes with loose caps. | Colpoda, Anophryoides, Chilodonella, Homalogastra, Pseudocohnilembus, Platyophrya, Opisthonecta |
| Dinoflagellates | Axenic culture grown in flasks or tubes. | Axenic strains of Crypthecodinium, Amphidinium |
| Free-living Flagellates | Aerobic xenic culture grown in flasks. | Xenic strains of Cercomonas, Ancyromonas, Bodo |
| Free-living Flagellates | Aerobic axenic culture grown in flasks. | Axenic strains of Diplonema, Rhynchopus |
| Free-living Flagellates | Microaerophilic, xenic culture grown in angled tubes. | Xenic strains of Hexamita, Mastigamoeba, Mastigella, Trepomonas |
| Non-motile Anaerobic Parasites | Non-motile parasites in xenic or axenic culture, grown anaerobically in tubes. | Blastocystis strains only |
| Non-Motile Parasites/ Saprophytes | Non-motile, non-photosynthetic parasites or saprophytes in axenic culture. Strains are grown in flasks. | Perkinsus, various thraustochytrids, Helicosporidium |
| Obligate Intracellular Parasites | Parasites grown in cell culture free from bacte1ria. Cultures are grown in flasks in a 5% CO2 atmosphere. | In vitro strains of Toxoplasma, Neospora, Encephalitozoon |
| Obligate Intracellular Parasites | Parasites grown in vivo. | Babesia microti, B. duncani, in vivo strains of Trypanosoma brucei, T. cruzi, Leishmania sp. |
| Parasitic Amoebae | Microaerophilic, parasitic strains in xenic or axenic culture. Strains are grown in angled tubes. | Entamoeba, Dientamoeba |
| Parasitic Flagellates | Parasitic flagellates in monoxenic or axenic, bacteria-free culture. Protists are grown upright in tubes or in some cases within flasks. A small number of strains are grown in cell culture within flasks at a 5% CO2 atmosphere. | Leishmania, Trypanosoma, Crithidia, Leptomonas, Herpetomonas, Phytomonas |
| Parasitic Flagellates | Microaerophilic, parasitic flagellates in axenic culture, grown in angled tubes. | Trichomonas, Tritrichomonas, Pentatrichomonas |
*Note that the examples included on this table are a list of the most common genera that fall into each category. The listed genera do not represent a complete accounting of the full taxonomic diversity of each type of culture. Further, the “protist culture type” is an internal ATCC categorization used for the purpose of generalizing culture protocols and handling techniques for similar organisms.
Protist metabolism
In general, protists have three methods of nutrient acquisition: autotrophy, heterotrophy, and mixotrophy. Autotrophic organisms, such as algae, require sunlight to obtain energy and organic material for carbon acquisition. These protist species use chlorophyll and various other pigments to obtain energy through photosynthesis. In contrast, heterotrophic organisms require the consumption of nutrients from other organisms for both energy and carbon. There are two forms of heterotrophy, phagotrophy, which is the engulfment of matter, and osmotrophy, which is the absorption of dissolved nutrients through pinocytosis. For example, free-living amoebae, such as Naegleria, directly consume bacteria through phagocytosis; whereas fungal-like protists, known as slime molds, consume organic matter by absorbing it from the environment. Lastly, mixotrophic organisms, such as most dinoflagellates, can use either, or both, autotrophic and heterotrophic forms of nutrient acquisition.
Protist propagation
The following section includes general guidelines for the culture of algae, bacteriovorus protists, and obligate intracellular parasites.
Algae
Image of Prototheca filamenta alga courtesy of Dr. W. Kaplan, M.S. Sudman, CDC
Algae are a very large and diverse group of autotrophic protists that range in size from unicellular microalgae to large multicellular organisms. To survive, algal species use chlorophyll and other pigments to manufacture their own food source through a process termed photosynthesis. The types of pigments used for photosynthesis differ among the algal divisions, resulting in colorful organisms that can appear green, red, brown, yellow, and golden-brown. In addition to variations in pigmentation, algal species also differ in the type of carbohydrate compounds stored for energy and the type of flagella used for motility. These differences can be used to help classify algal species.
In the laboratory, algae are commonly grown as axenic cultures in either liquid media or on agar media. Strains are commonly propagated in aerobic/ventilated culture vessels under a 14 hour light/10 hour dark cycle to promote photosynthesis. ATCC recommends the following procedure for the in vitro cultivation of algae:
Propagation
- Rehydrate freeze-dried preparations by aseptically adding 0.5 mL of ice-cold medium to the freeze-dried inner shell vial. For frozen preparations, thaw the contents of the ampoule by placing the vial in a 35°C water bath until thawed (approximately 2 minutes). Immerse the ampoule just sufficiently to cover the material. Do not agitate the ampoule.
- Aseptically transfer the entire contents to a 16 × 125 mm screw-capped test tube containing 5 mL of the recommended medium. Alternatively, add the entire contents to the surface of a 20 × 100 mm Petri plate containing 20 mL of the recommended medium. Some algae, and particularly freeze-dried preparations, do not recover well in broth media. For these cultures, agar media is recommended.
- Incubate broth cultures horizontally at a 15° slant with the cap screwed on loosely at 25°C under a 14 hour light /10 hour dark cycle. For agar-based cultures, wrap the plate culture in Parafilm and incubate upright.
- Subculture as necessary. This may be every 14-21 days in broth media (See: NOTE 3).
Note 3:
Some green algae may lose their color shortly after recovery from the frozen state. In most cases, the natural coloration will return after a period of time with normal incubation.
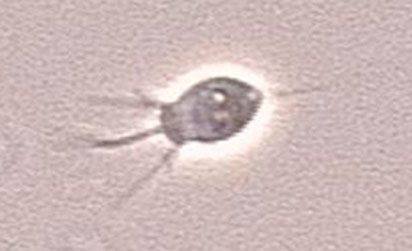
Bacterivorous protists
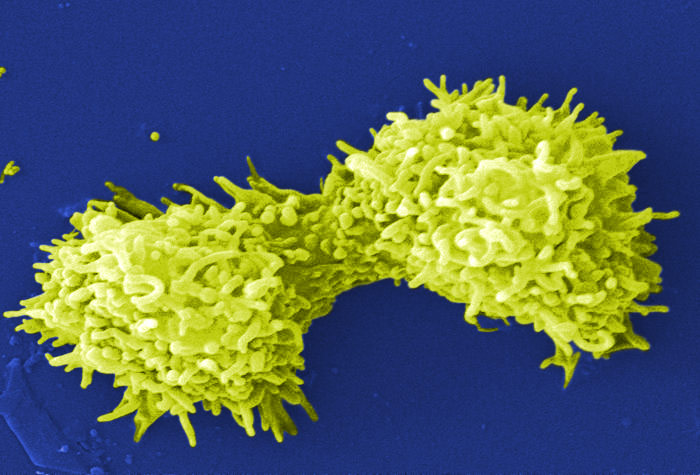
Image of Acanthamoeba polyphaga. Photo courtesy of Catherine Armbruster and Margaret Williams.
Xenic, bacterized protist cultures require the presence of bacteria as a food source. Typically, the bacterial strain will survive the preservation process better than most protists, and will be present in the culture when it is re-initiated from preserved material. The concentration of the bacterial food source can affect the growth of many protist species or may make it difficult to observe the protist strain. For these cultures, control over bacterial density may enhance protist propagation. The following procedures regarding propagation and bacterial control can be applied to the majority of free-living, aerotolerant, xenic protist cultures:
Propagation
- Rehydrate freeze-dried preparations by aseptically adding 0.5 mL of ice-cold medium to the freeze-dried inner shell vial. For frozen preparations, thaw the contents of the ampoule by placing the vial in a 35°C water bath until thawed (approximately 2 minutes). Immerse the ampoule just sufficiently to cover the material. Do not agitate the ampoule.
- Transfer the entire contents to 10 mL of the appropriate ATCC medium in a T-25 tissue culture flask. The bacterial flora present in the shipped culture will support growth, but growth may be improved by using medium that has been bacterized with Enterobacter aerogenes approximately 24 hours before inoculation with the protist (See: NOTE 4, Table 3).
- Subculture every 14-21 days. To subculture, aseptically scrape the bottom of the flask and agitate the flask to distribute the cells evenly. Aseptically transfer 0.25 to 0.5 mL to a T-25 flask containing 9 mL of bacterized medium.
- Incubate the flask at 25°C with the cap screwed on tightly.
Note 4:
All three bacterial strains listed in Table 3 can be used almost interchangeably with the vast majority of free-living, bacteriovorus protist strains
Table 3. Bacterial strains used as a food-source for free-living bacteriovorus protists
| ATCC number | Species |
|---|---|
| 27889 | Klebsiella pneumoniae subsp. pneumoniae |
| 13048 | Enterobacter aerogenes |
| 700831 | Klebsiella pneumoniae subsp. pneumoniae |
For some xenic protist cultures, better growth may be obtained if bacterial density is reduced to some degree. This phenomenon is not uncommon among aerobic, free-living flagellate, ciliate, and amoeba cultures, particularly where mixed bacteria flora are present. Microaerophilic protists in xenic culture may also exhibit reduced or inhibited growth if bacterial density becomes excessive, primarily as a result of mixed bacterial flora native to the culture. Some degree of control over bacterial density in these cultures can be exerted through the addition of antibiotics or by a reduction in the quantity or strength of whichever component in the growth medium is promoting excessive bacterial growth. Any alteration to growth media should be tested empirically to determine the optimal culture conditions.
Controlling bacterial density in xenic cultures
- Dilute Cerophyl media to reduce bacterial density by mixing it 1:1 or 1:2 with a suitable buffer medium, such as ATCC media formulations 2348 or 1323 for freshwater cultures or ATCC medium formulation 1405 for marine cultures (See: NOTE 5).
- Media containing rice starch can sometimes promote excessively dense bacterial growth. Manual reduction in the amount of rice starch in the medium prior to inoculation with bacteria can help modulate subsequent bacterial growth. Rice starch may be removed from media by light centrifugation.
- Further control of bacterial growth may require use of antibiotics such as penicillin/streptomycin; however, the appropriate concentrations should be determined empirically.
Note 5:
ATCC medium formulations can be found on the ATCC website.
Free-living amoebae such as Naegleria, Hartmannella, or Acanthamoeba in xenic/bacterized culture on agar media covered with dense bacterial growth can be challenging to observe or cultivate successfully. Especially for the inexperienced, it can be quite difficult to discern amoebae amidst the dense growth of mixed bacterial flora. To achieve optimal culture conditions, it can be helpful to either reduce the overall bacterial density or to separate amoebae from the dense bacteria around them.
Reducing bacterial density on agar media
An amoeba culture containing mixed bacterial flora may be washed with a suitable buffer solution to remove most of the bacteria, potentially enabling remaining amoebae to grow to greater numbers.
- Flood a 20 × 100 mm agar petri plate containing the amoeba culture with 4 to 5 mL of a balanced saline solution. For freshwater strains, use Page's amoeba saline (ATCC medium formulation 1323) or PBS; for marine strains, use an artificial seawater wash.
- Gently rub the plate surface with a spread bar to carefully dislodge adherent amoebae, and then transfer suspended amoebae and bacteria to a 15 mL plastic centrifuge tube.
- Centrifuge the culture at approximately 400 to 500 × g for 5 to 6 minutes.
- Remove all but 0.5 to 1.0 mL of the supernatant and then resuspend pelleted amoebae. Make a wet mount slide to confirm presence of amoebae or cysts. Transfer the resuspended culture material onto several fresh, uninoculated agar plates and spread over the plate surface using a spread bar.
- Allow all plates to air dry, then seal with parafilm and incubate as recommended. Observe cultures daily for presence of newly-emergent amoebae; these may be further isolated from problematic bacterial growth as described below.
Separating amoebae from undesirable bacteria on agar media
A fresh lawn of a single, preferred bacterial species may be less dense than the mixed bacterial flora on the parent culture plate, offering a better opportunity for amoebae to proliferate in a more visible way.
- Prepare a lawn of either E. aerogenes or K. pneumoniae bacteria on several fresh agar plates, and allow the plates to dry completely.
- Transfer the amoeba culture with an inoculation loop to a location on one of the freshly-prepared agar plates containing the lawn of a single bacterial species. Attempt to transfer from a region on the source plate that contains amoeba tophozoites or cysts if possible; otherwise, if amoebae or cysts are not apparent, blind transfers can be made to multiple subculture plates (See: NOTE 6).
- Amoebae that successfully transfer to a new agar plate may proliferate more readily and subsequently migrate to a region of the plate containing only the preferred bacterial species. A second transfer of amoebae from this new region will result in a monoxenic culture of amoebae and a single bacterial food source when inoculated onto a fresh agar plate pre-inoculated with a lawn of the preferred bacterium.
Note 6:
Instead of using an inoculating loop to transfer cells, a small block of agar may be cut from the parent culture plate using a sterilized fine-point scalpel or Pasteur pipette tip. The agar block is then physically transferred to one of the freshly-prepared subculture plates and inverted onto the surface of the bacterial lawn. Gently slide the agar block along the lawn a short distance to transfer amoebae from the block to the new bacterial lawn.
Intracellular parasites
Several protist species, such as Toxoplasma gondii and the fungus Encephalitozoon cuniculi, are considered obligate intracellular parasites. These organisms require a living host cell for in vitro propagation. Below are general ATCC procedures that describe how to prepare a host cell line and propagate an intracellular parasite culture (See: NOTE 7, Table 4).
Note 7:
All procedures pertaining to the manipulation of cells and dispensing of growth media into flasks should be performed in a biological safety cabinet.
Table 4. Cell lines used in the in vitro propagation of intracellular parasites
| ATCC number | Description | Application |
|---|---|---|
| CCL-34 | MDCK | Encephalitozoon sp., Vittaforma corneae |
| CCL-37 | RK13 | Encephalitozoon sp. |
| CCL-75 | Human lung fibroblasts | Encephalitozoon sp. |
| CCL-81 | Vero | Toxoplasma gondii, Encephalitozoon sp., Trypanosoma cruzi |
| CCL-163 | BALB/3T3 fibroblasts | Trypanosoma cruzi |
| CRL-1634 | Human foreskin fibroblasts | Toxoplasma gondii |
In vitro propagation
Preparation of the host cell line
- Thaw a vial of the host cell culture by placing the ampoule in a water bath set at 35°C (approximately 2 minutes). Immerse the vial just sufficiently to cover the frozen material. Do not agitate the vial.
- Sterilize the outer surface of the vial using freshly prepared 70% ethanol.
- Aseptically transfer the entire thawed contents of the ampoule to a T-25 tissue culture flask containing 10.0 mL of the recommended medium. Be sure that your medium is properly buffered with sodium bicarbonate to ensure proper cellular growth in a 5% CO2 incubator.
- If flasks with plug seal caps are used, outgas each flask for 30-45 seconds with a 95% air, 5% CO2 gas mixture. Tightly cap the flask.
- Incubate the flask horizontally in a in a 35-37°C, 5% CO2 incubator.
- Observe the culture regularly using an inverted microscope to determine the health and confluency of the cells. When the cells form a monolayer completely covering the bottom surface of the flask, the culture may be passaged or expanded.
Transferring the host cell line
- When the cell line reaches the desired density, remove the medium and replace it with an appropriate volume of Phosphate Buffered Saline (PBS) (ATCC 30-2200) to cover the cell layer. Incubate the flask at 35°C to 37°C for 3-5 minutes (or up to 30 minutes for cell lines that do not detach easily) to remove serum and/or other potential trypsin inhibitors present.
- Remove all the PBS and replace it with a small volume of 1X Trypsin-EDTA solution (ATCC 30-2101) sufficient to leave a thin film of the solution over the entire bottom surface of the flask. For a T-25 flask, 1-2 mL of 1X Trypsin-EDTA solution should be sufficient (See: NOTE 8).
- Incubate the flask horizontally at 35°C to 37°C until the desired detachment results are obtained. Cells may be observed using an inverted microscope to confirm they have become rounded and detached.
- Neutralize the Trypsin-EDTA solution by dispensing an appropriate volume of the growth medium with FBS. For medium with 10% FBS, add at least 5 mL for every 1 mL of trypsin used. For medium with 5% FBS, add at least 10 mL for every 1 mL of trypsin used.
- Resuspend the cells by aspirating and expelling the fluid gently with a pipette and transfer an appropriate volume to each of the desired number of destination flasks.
- Aseptically dispense suitable fresh growth medium with the defined concentrations of supplements to each destination flask at an appropriate volume. If flasks with plug seal caps are used, outgas each destination flask for 30-45 seconds with a 95% air, 5% CO2 gas mixture and tightly cap the flask.
- Incubate the flask horizontally at 35-37°C in a 5% CO2 incubator.
Note 8:
Trypsin-EDTA solution must be fresh for proper detachment. Some cell lines will not detach easily or completely, in which case the addition of other enzymes may be necessary.
Propagation
- Thaw a frozen ampoule containing the protist strain by placing it in a 35°C water bath such that the lip of the ampoule remains above the water line. Thawing time is approximately 2 minutes. Do not agitate the ampoule. Do not leave ampoule in water bath after it is thawed. Further, protist cultures should not be thawed at room temperature or in the refrigerator.
- Aseptically transfer the entire thawed contents of the ampoule to a T-25 tissue culture flask containing a fresh monolayer of the propagation host cells and 10 mL of the recommended medium.
- Incubate in a 35-37°C, 5% CO2 incubator. If a flask with a plug seal cap is used, outgas the flask for 10 seconds with a 95% air, 5% CO2 gas mixture. Tightly cap the flask.
- Observe the culture daily under an inverted microscope for the presence of intracellular parasites inside the cells.
- To maintain the culture, remove the medium from a fresh confluent monolayer of the host cell line in a T-25 tissue culture flask and replace it with 10 mL of the recommended medium.
- Transfer the protist culture by removing the old medium from the flask containing the organism and centrifuge at 1300 × g for 10 min.
- Remove all but 0.5 mL of the supernatant and resuspend the cell pellet. Transfer the resuspended pellet to the fresh flask containing the host cell line. Outgas the flask for 10 seconds with a 95% air, 5% CO2 gas mixture if using a plug seal cap.
- Incubate at 35-37°C in a 5% CO2 incubator with the cap screwed on tightly.
Parasites requiring in vivo propagation
For the propagation of parasitic protists that lack an in vitro culture system such as certain species of Babesia, in vivo cultivation using an appropriate host is required. The required host species and inoculation route can vary significantly depending on the species. Below is a general ATCC procedure for in vivo cultivation of parasites. For specific details regarding the recommended preparation of a protist strain and the method of inoculation, please review the product sheet for that organism.
Propagation
- Prior to thawing the frozen ampoule, prepare the host animal under the recommended conditions.
- Once the host animal is prepared, thaw the frozen ampoule in a 35°C water bath such that the lip of the ampoule remains above the water line. Thawing time is approximately 2 minutes. Do not agitate the ampoule. Do not leave ampoule in water bath after it is thawed. Further, protist cultures should not be thawed at room temperature or in the refrigerator.
- Immediately after thawing, aseptically remove the contents of the ampoule with a syringe and inoculate an uninfected, animal host. Follow the recommended protocol for maintenance in vivo described on the product sheet; this procedure will vary significantly between protist species. Note that the course of infection may also vary depending on the virulence of the parasite strain and its recovery from the frozen state.
- Monitor parasitemia and passage as needed.
Propagation of specific protist species
The following section includes general guidelines for the culture of some of the most commonly ordered protist species from ATCC. Additional information regarding the recommended media used in the cultivation of these species is found on the ATCC website as well as on the product sheet.
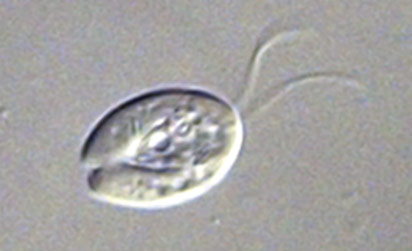
Trichomonas vaginalis
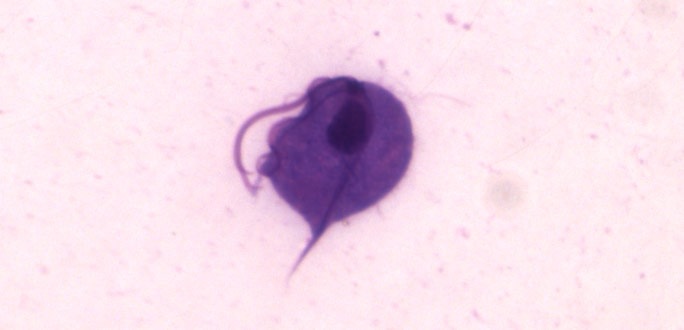
Image of Trichomonas vaginalis.
T. vaginalis is a single-celled, flagellated protozoan known to cause the human sexually transmitted disease, trichomoniasis. This protist is oval in shape with four posterior flagella and a fifth flagellum wrapped around the surface of the organism. A conserved feature of T. vaginalis is the production of adherence factors that allow for epithelial attachment and colonization. ATCC recommends the following procedure for the propagation of T. vaginalis:
- Thaw a frozen ampoule by placing it in a 35°C water bath until thawed (approximately 2 minutes). Immerse the ampoule just sufficiently to cover the frozen material. Do not agitate the ampoule.
- Immediately after thawing, aseptically transfer the entire contents to a screw-capped test tube containing either 9.0 mL of completed TYM media adjusted to pH 6.0 or 13.0 mL of completed LYI media adjusted to pH 6.0. Incubate the tube at 35°C in the upright position in the case of TYM media, or horizontally at a 15° slant in the case of LYI media.
- When the culture reaches peak density, place the tube on ice for 10 minutes.
- Gently invert the culture tube 10 times and aseptically transfer a 0.1-0.4 mL aliquot to a screw-capped test tube containing an appropriate volume of the recommended medium as described in step 2.
- Screw the cap on tightly and incubate the culture at 35°C.
- Transfer the culture every 3-4 days as described in steps 3-5. The transfer interval will depend on the quantity of the inoculum and the quality of the medium. This should be empirically determined by examining the culture on a daily basis, until the growth cycle has stabilized. Do not allow the culture to overgrow as protist death occurs soon after reaching peak density.
Acanthamoeba castellanii
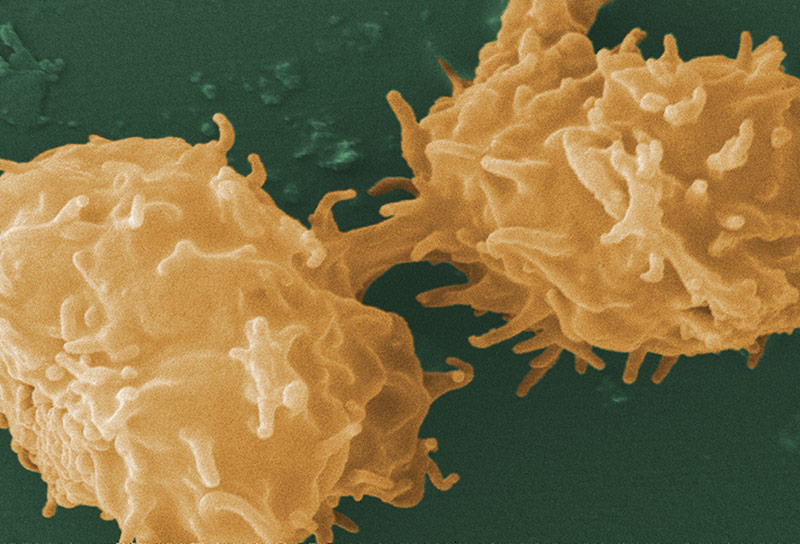
Image of Acanthamoeba sp. courtesy of Janice Haney Carr, CDC
A. castellanii is a freshwater amoeba known to cause keratitis and encephalitis in humans. Cells are small, can form cysts, and are oval to triangular in shape. In addition to their role as a human pathogen, A. castellanii is also a host to various bacterial strains, including Legionella pneumophila. ATCC recommends the following procedure for the propagation of axenic strains of A. castellanii:
- To thaw a frozen ampoule, place it in a 35°C water bath such that the lip of the ampoule remains above the water line. Thawing time is approximately 2 minutes. Do not agitate the ampoule. Do not leave ampoule in water bath once thawed. Protists should not be thawed at room temperature or in the refrigerator.
- Immediately after thawing, aseptically transfer the entire contents to either a 16 × 125 mm plastic test tube or a T-25 tissue culture flask containing the recommended medium (5.0 mL in a test tube, 10.0 mL in a flask).
- Screw the cap on tightly and incubate the tube or flask at 25°C.
- When the culture reaches peak density, vigorously agitate the culture.
- Transfer approximately 0.25 mL to a fresh tube or flask of the recommended medium, freshly prepared (5.0 mL in a test tube, 10.0 mL in a flask).
- Screw the cap on tightly and incubate at 25°C (incubate test tubes horizontally at a 15° slant).
- The amoebae will form an almost continuous sheet of cells on the bottom surface of the flask or test tube. Repeat steps 4-6 at 10-14 days intervals.
For propagation of xenic/bacterized A. castellanii strains, ATCC recommends the following:
- To thaw a frozen ampoule, place it in a 35°C water bath such that the lip of the ampoule remains above the water line. Thawing time is approximately 2 minutes. Do not agitate the ampoule. Do not leave ampoule in water bath once thawed. Protists should not be thawed at room temperature or in the refrigerator.
- Immediately after thawing, aseptically transfer the entire contents to an agar plate of the recommended medium. Distribute the material evenly over the plate using a spread bar and allow to dry.
- Wrap the entire edge of the plate with parafilm and incubate upright at 25°C. Amoeba trophozoites should be evident within 2-5 days.
- When the amoeba culture reaches peak density, transfer amoebae or cysts to a fresh plate of the recommended medium with an inoculation loop. Agar plates pre-inoculated with a lawn of food-source bacteria will promote faster growth of amoebae.
- Seal the agar plate with parafilm and incubate upright at 25°C.
- The amoebae will form an almost continuous sheet of cells on the surface of the agar. Repeat steps 4-5 at 7-10 day intervals.
Giardia intestinalis
G. intestinalis is a flagellated protozoan that is known to cause giardiasis in humans and other mammals. These organisms enter the host intestine through the consumption of soil, food, or water that has been contaminated with the feces of an infected individual. During infection, G. intestinalis attaches to the intestinal epithelium and reproduces by binary fission. ATCC recommends the following procedure for the propagation of G. intestinalis:
- To thaw a frozen ampoule, place it in a 35°C water bath until thawed (approximately 2 minutes). Immerse the ampoule just sufficiently to cover the frozen material. Do not agitate the ampoule. Protists should not be thawed at room temperature or in the refrigerator.
- Immediately after thawing, aseptically transfer the entire contents to a screw-capped test tube containing 13 mL of medium. Commonly use media include Keister's Modified TYI-S-33 (ATCC medium 2695) and LYI Giardia medium (ATCC medium 2155). Incubate the tube on a 15° horizontal slant at 35°C.
- When the culture has reached or is near peak density, place the tubes on ice for 10 minutes.
- Gently invert the culture tube 10 times and aseptically transfer a 0.1-0.4 mL aliquot to a screw-capped test tube containing 13 mL of the recommended medium.
- Incubate the culture horizontally at a 15° slant at 35°C with the cap on tightly.
- Transfer the culture every 3-4 days as described in steps 3-5. The transfer interval will depend on the size of the inoculum and the quality of the medium. This should be empirically determined by examining the culture on a daily basis until the growth cycle has stabilized. Do not allow the culture to overgrow. The culture crashes soon after reaching peak density.
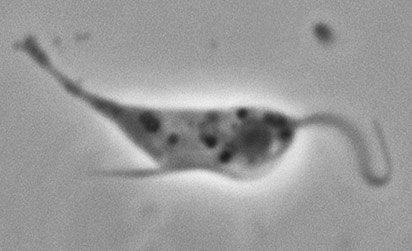
Trypanosoma cruzi
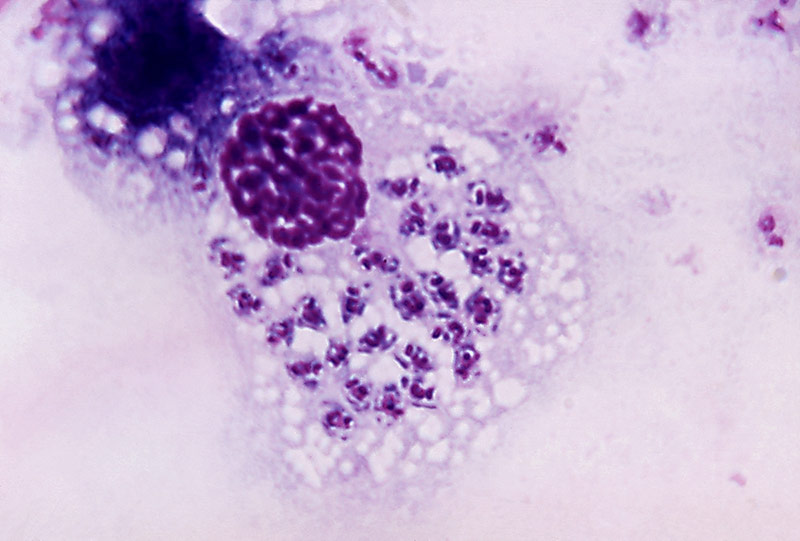
Image of Trypanosoma cruzi courtesy of Dr. A.J. Sulzer, CDC
T. cruzi is a vector-borne parasitic euglenoid trypanosome that causes Chagas disease in humans. Within the human body, T. cruzi can be found in two forms: The trypomastigote, which is the infectious stage in the human bloodstream, and the amastigote, which is the reproductive stage that occurs within host cells. The epimastogote stage is found naturally in the insect vector and is the life stage most commonly cultured in vitro. ATCC recommends the following procedure for the propagation of T. cruzi strains adapted to in vitro cultivation in LIT medium:
- To thaw a frozen ampoule, place it in a 35°C water bath until thawed (approximately 2 minutes). Immerse the ampoule just sufficiently to cover the frozen material. Do not agitate the ampoule. Protist cultures should not be thawed at room temperature or in the refrigerator.
- Immediately after thawing, aseptically transfer the entire contents to a T-25 tissue culture flask containing 10 mL of medium. Incubate the tube at 25°C.
- When the culture is at or near peak density, agitate the flask by inverting several times.
- Aseptically transfer a 0.1 mL aliquot to a T-25 tissue culture flask containing 10 mL of the recommended medium, freshly prepared.
- Screw the cap on tightly and incubate the flask at 20°C to 25°C.
- Subculture every 7-14 days.
Entamoeba histolytica
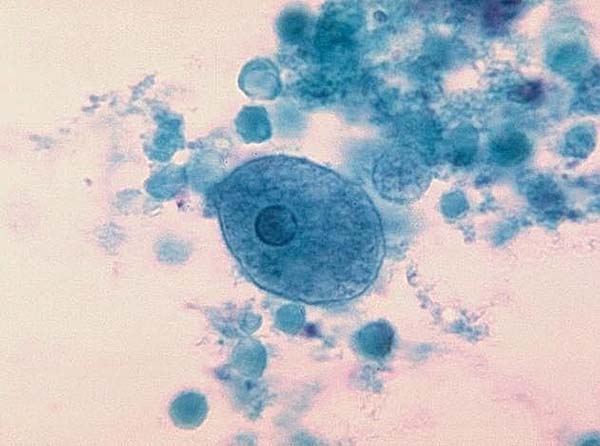
Image of Entamoeba histolytica courtesy of Dr. George Healy, CDC
E. histolytica is a parasitic protozoan that is transmitted among humans and primates through the consumption of contaminated water, solids, and foods. Upon ingestion of E. histolytica cysts, amoeba trophozoites are extruded into the digestive tract, eventually causing amoebic dysentery or the formation of amoebic liver abscesses. ATCC recommends the following procedure for the propagation of axenic strains of E. histolytica that are distributed as frozen preparations:
- To thaw a frozen ampoule, place in a 35°C water bath until thawed (approximately 2 minutes). Immerse the ampoule just sufficiently to cover the frozen material. Do not agitate the ampoule. Protist cultures should not be thawed at room temperature or in the refrigerator.
- Immediately after thawing, aseptically transfer the entire contents to a glass screw-capped tube containing 13 mL of medium. Axenic strains are commonly cultured in LYI medium (ATCC medium 2154) while xenic strains are cultured in TYGM-9 medium (ATCC medium 2154, 2463, and 2692). Screw the cap on tightly and incubate horizontally at a 15° slant at 35°C.
- When the culture is at or near peak density, ice the culture tube for 10 min.
- Gently invert culture 20 times to dislodge adherent amoeba.
- Aseptically transfer a 0.1 and 0.25 mL aliquot to freshly prepared (no older than 7-10 days) tubes of the recommended medium.
- Screw caps on tightly and incubate horizontally at a 15° slant at 35°C.
- Subculture as described in steps 3-6 when many trophozoites are observed (typically every 2-4 days). The transfer interval will depend on the quantity of the inoculum and the quality of the medium. This should be empirically determined by examining the culture on a daily basis until the growth cycle has stabilized. Do not allow the culture to overgrow. The culture crashes soon after reaching peak density.
For propagation of xenic/bacterized E. histolytica strains distributed as growing cultures, ATCC recommends the following:
- Upon receipt, immediately incubate culture horizontally at a 15° slant at 35°C for at least three hours before observing the culture. Amoeba trophozoites should be evident attached to the tube wall amidst rice starch and bacteria.
- Regardless of the state of the culture, remove 5.5 mL of fluid and centrifuge at 500 × g for 5 minutes.
- Inoculate 2 fresh tubes of the recommended medium with 0.25 mL of supernatant (preinoculated bacterized culture tubes allow for better growth) (See: NOTE 9). Xenic strains are commonly cultured in 8 mL TYGM-9 medium (ATCC medium 1171).
- Divide the remainder of the supernatant in two equal aliquots in 16 × 125 mm screw-capped tubes and bring the volumes up to 8 mL with fresh medium.
- Ice the parent culture 5 minutes, invert 20 times and transfer 0.5 and 1.0 mL aliquots to the test tubes containing the equal volumes of supernatant.
- Re-feed the parent culture by centrifuging it at 200 × g for 5 min, aspirate most of the supernatant (leaving approximately 1.0-1.5 mL), and resuspend the pellet with fresh growth medium up to 8 mL.
- Incubate all cultures (including the tubes of bacterized growth medium) horizontally at a 15° slant at 35°C.
- Observe the culture daily and transfer when many trophozoites are observed (ie, early stationary phase): Ice a test tube culture at or near peak density for 10 minutes, invert 20 times and aseptically transfer a 0.1 and 0.3 mL aliquot to a fresh tube of recommended medium.
- Screw cap on tightly and incubate horizontally at a 15° slant at 35°C.
Note 9:
Growth of xenic/bacterized E. histolytica is often better if the growth medium is inoculated with bacteria at least 24 hrs before inoculating with amoebae. Also, growth can be improved by controlling bacterial density with addition of antibiotics to the culture. In general, addition of penicillin G at 75 U/mL and streptomycin at 75 µg/mL to the medium may be necessary if the bacterial density in the culture is excessive. Antibiotics, if used, should be added at least 24 hours after medium is inoculated with bacteria.
Babesia microti
B. microti is a blood parasite that causes the hemolytic disease, babesiosis. This organism is spread between hosts through the saliva of the Ixodes scapularis tick. Upon infection, B. microti enters erythrocytes where it is able to develop and eventually reproduce. ATCC recommends the following procedure for the in vivo cultivation of B. microti:
- To thaw a frozen ampoule, place it in a 35°C water bath such that the lip of the ampoule remains above the water line. Thawing time is approximately 2 minutes. Do not agitate the ampoule. Do not leave ampoule in water bath after it is thawed. Further, protist cultures should not be thawed at room temperature or in the refrigerator.
- Immediately after thawing, aseptically inoculate entire infected blood suspension intraperitoneally into a hamster using a 1.0 mL syringe equipped with a 27 gauge 1/2 inch needle. Note that the course of infection may vary depending on the virulence of the parasite strain and its recovery from the frozen state (See: NOTE 10).
- Monitor the infection at 1-2 day intervals by examination of blood films stained with 5% Giemsa solution.
- Count the number of infected red blood cells (RBC) versus the total number of red cells under oil immersion and determine the % parasitemia: % parasitemia = infected rbc / total rbc × 100. A minimum of 500 red blood cells should be counted. (Note that a red blood cell infected with multiple parasites is counted as a single infected cell.)
- When the level of parasitemia is ≥ 10% the strain should be passaged. Normally this would occur 1-3 weeks post-inoculation, but the rate of infection may vary considerably. (Note that the level of parasitemia before the host will succumb will vary with the strain used. Monitoring on a daily basis will alert the researcher as to when the strain should be passaged.)
- Passage the strain by removing blood from the infected hamster using an appropriate cardiac puncture procedure.
- Inject 0.5 mL of the infected blood suspension into each uninfected hamster.
- Monitor parasitemia and passage as needed.
Note 10:
Hamsters may be primed for faster infection by treatment with Cortisone (2mg/day/hamster) 1-3 days prior to inoculation.
Toxoplasma gondii
T. gondii is an obligate, intracellular protozoan known to cause the disease toxoplasmosis. Infection in humans and mammals can occur by the consumption of raw or undercooked meat containing T. gondii cysts, through the ingestion of products contaminated with oocysts, or by transplacental transmission. ATCC recommends the following procedure for the propagation of T. gondii:
Preparation of the host cell line
- To establish a cell culture from the frozen state place an ampoule in a water bath set at 35°C (2-3 min). Immerse the vial just sufficiently to cover the frozen material. Do not agitate the vial.
- Immediately after thawing, aseptically remove the contents of the ampoule and inoculate into 10.0 mL of fresh media in a T-25 tissue culture flask.
- Incubate in a 35-37°C, 5% CO2 incubator. If a flask with a plug seal cap is used, outgas the flask for 10 seconds with a 95% air, 5% CO2 gas mixture. Tightly cap the flask.
- Change the medium 1-2 times per week.
Transferring the host cell line
- When the cell line forms a confluent layer, remove the medium and replace it with 3 mL of Phosphate Buffered Saline (PBS) (ATCC 30-2200). Incubate the flask at 35-37°C for 3-5 minutes (or up to 30 minutes for cell lines that do not detach easily) to remove serum and/or other potential trypsin inhibitors present.
- Remove all the PBS and replace it with 2 mL of 0.25% (w/v) trypsin dissolved in Hank's Balanced Salt Solution (ATCC 30-2101).
- Gently distribute the trypsin over the monolayer, remove the trypsin, and place the flask at 35-37°C for 5-10 minutes or until the desired detachment results are obtained.
- Add 2 mL of the recommended growth medium and detach any cells still adherent by alternately aspirating the medium into a pipette and discharging the contents over the monolayer.
- Distribute the cell suspension in 0.5 mL aliquots to 4 T-25 flasks containing 10 mL fresh growth medium.
- Incubate in a 35-37°C, 5% CO2 incubator. If a flask with a plug seal cap is used, outgas the flask for 10 seconds with a 95% air, 5% CO2 gas mixture. Tightly cap the flask.
Propagation
- Thaw a frozen ampoule by placing it in a 35°C water bath such that the lip of the ampoule remains above the water line. Thawing time is approximately 2 to 3 minutes. Do not agitate the ampoule. Do not leave ampoule in water bath after it is thawed.
- Aseptically transfer the entire thawed contents of the ampoule to a T-25 tissue culture flask containing a fresh monolayer of the propagation host cells and 10 mL of the recommended medium.
- Incubate in a 35-37°C, 5% CO2 incubator. If a flask with a plug seal cap is used, outgas the flask for 10 seconds with a 95% air, 5% CO2 gas mixture. Tightly cap the flask.
- Observe the culture daily for the presence of intracellular parasites inside the cells.
- To maintain the culture, remove the medium from a fresh confluent monolayer of the host cell line in a T-25 tissue culture flask and replace it with 10 mL of the recommended medium.
- Transfer the Toxoplasma culture by removing the old medium from the flask, and centrifuge at 1300 × g for 10 min.
- Remove all but 0.5 mL of the supernatant and resuspend the cell pellet. Transfer the resuspended pellet to the fresh flask containing the host cell line. Outgas the flask for 10 seconds with a 95% air, 5% CO2 gas mixture if using a plug seal cap.
- Incubate in a 35-37°C CO2 incubator with the cap screwed on tightly.
Cell counts
Cell counts are often necessary to monitor growth rates of protists and establish new cultures. Hemocytometers (also spelled hemacytometers) are commonly used to estimate protist cell density and determine viability. A hemocytometer is a thick glass slide with two counting chambers, one on each side. Each counting chamber has a mirrored surface with a 3 × 3 mm grid of 9 counting squares. The chambers have raised sides that will hold a coverslip exactly 0.1 mm above the chamber floor. Each of the 9 counting squares holds a volume of 0.0001 mL.
Hemocytometers are excellent for determining culture viability and estimating density, but are not precise for determining actual cell numbers due to the relatively low number of cells actually counted. Although rarely used with protist cultures, an automated counter will generate the most reliable data, particularly when used in combination with the viability data from a hemocytometer.
Count cells as follows:
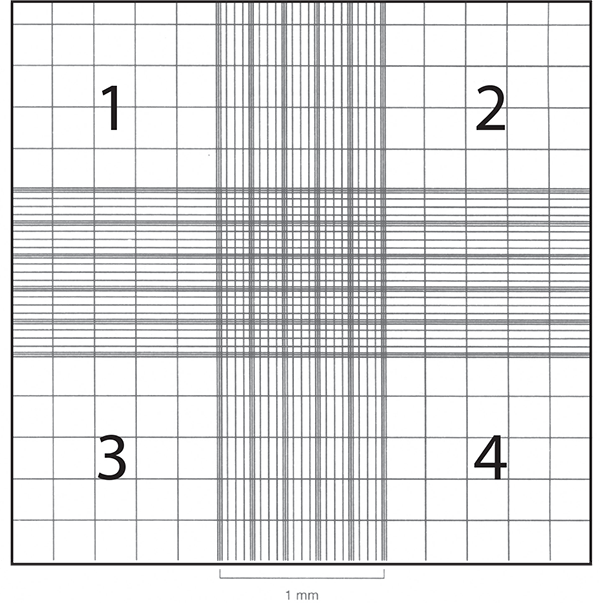
- Clean, thoroughly dry, and assemble the hemocytometer with the cover slip.
- Transfer a small amount of the culture suspension to the edge of each of the two counting chambers. Allow the culture to be drawn into the counting chamber by capillary action.
- Place the hemocytometer over the objectives of an inverted microscope and view the cells at 100× magnification.
- Focus on the squares of each of the four corners, labeled 1, 2, 3, and 4 in the adjacent figure.
- Record the number of cells in each square. Average the number of cells, and multiply by the dilution factor. If the cells have not been diluted, this factor will be 104 cells/mL. Any dilution of the sample after it was removed from the cell suspension, such as using vital stain, needs to be included in the calculation.
For example, if the four counts are 60, 66, 69, and 75, the concentration would be 68 × 104 protists/mL for the sample that was loaded into the hemocytometer. For best results, adjust the concentration of the suspension so that 50 to 100 cells are in each of the four counting squares.
The growth rate and culturing requirements of protists can vary drastically between species. ATCC recommends that growth rates be determined empirically. For more information on how to culture a particular strain, see the product sheet for details or contact ATCC Product Experience.
Growth media
Protist culture media
Protist culture media are often mixtures of proteins, trace elements, amino acids, and carbohydrates. The requirements for these components vary among algal and protozoa strains. In addition to supplying nutrients, medium assists in the maintenance of pH. The pH can be sustained through one or more buffering systems such as MOPS or potassium phosphate. ATCC uses numerous types of media in order to provide the optimal growth conditions for each protist. Each medium is tested by ATCC for microbial growth promotion. The recommended media for all strains is indicated on the product sheet and online at www.atcc.org.
Keister’s Modified tYI-S-33 Giardia Medium (ATCC medium formulation 2695, ATCC PRA-2695) is a complex growth medium used for the axenic cultivation of Giardia and Spironucleus. This medium is prepared as a mixture of casein digest, yeast extract, bovine bile, salts, Diamond’s Vitamin Solution, and heat-inactivated bovine serum. It is provided freeze-dried, complete with 10% bovine serum.
LYI Entamoeba Medium (ATCC medium formulation 2154, ATCC PRA-2154) is a growth medium used for the axenic cultivation of Entamoeba, Hypotrichomonas, Mastigamoeba, Pentatrichomonas, Tetratrichomonas, Trichomitus, Trichomonas, and Tritrichomonas. This medium is prepared as a mixture of neutralized liver digest, yeast extract, glucose, salts, vitamins, and heat-inactivated bovine serum. It is provided freeze-dried, complete with 10% bovine serum.
LYI Giardia Medium (ATCC medium formulation 2155, ATCC PRA-2155) is a growth medium used for the axenic cultivation of Giardia and Spironucleus. This medium is prepared as a mixture of neutralized liver digest, yeast extract, glucose, salts, vitamins, and heat-inactivated bovine serum. It is provided freeze-dried, complete with 10% bovine serum.
Modified PYNFH Medium (ATCC medium formulation 1034, ATCC 327-X) is a growth medium used for the axenic cultivation of small, free-living amoebae including Hartmanella and Naegleria, as well as insect trypanosomes including Crithidia, Herpetomonas, Leptomonas, Phytomonas, and Tetrahymena. This medium is prepared as a mixture of peptone, yeast extract, yeast nucleic acids, folic acids, salts, and heat-inactivated fetal bovine serum. It is provided freeze-dried, complete with 10% fetal bovine serum.
TYGM-9 Medium (ATCC medium formulation 1171, ATCC PRA-1171) is a growth medium used for the cultivation of bacterized strains of Entamoeba, Dientamoeba, and Endolimax. This medium is prepared as a mixture of yeast extract, gastric mucin, salts, heat-inactivated bovine serum, tween 80, and rice starch. It is provided in liquid form, complete with 3% bovine serum.
Media formulations
The formulations for media used by ATCC can be found on the ATCC website at www.atcc.org. Please note that the majority of ATCC microbial media are not currently available for sale from ATCC.
Media ingredients
Diamond’s Vitamin Solution
Diamond’s Vitamin Solution is used to supplement protist growth media with vitamins and nutrients that stimulate growth, reduce the biosynthetic burden of in vitro cultures, and prolong cell viability.
Neutralized Liver Digest
A standardized digest of liver is used as a source of nutrients or as a supplement in microbial culture media. This reagent contains relatively high levels of iron and can be used as a source of nitrogen and amino acids.
Peptone
Peptone is a water-soluble protein derivative used in culture media. This reagent is prepared via the partial hydrolysis of an animal protein by an enzyme or acid. Not all microbial species can use free atmospheric nitrogen. Many species require either organic or inorganic fixed nitrogen.6 Peptone is used in media as an organic source of nitrogen and is often used in serum-free medium. The nutritional value of peptone is dependent on the amino acid content that supplies the essential nitrogen. The starting material for peptone can range from animal to plant; these can include meat, soybean, casein, and whey.7
Serum
Sera can serve as a source of growth factors, proteins, vitamins, hormones, carbohydrates, lipids, amino acids, minerals, and trace elements. The exact composition of sera is unknown and varies from lot to lot, although consistency from one lot to another has improved in recent years. Sera from bovine and fetal bovine sources are often used to support the growth of some protist strains in culture. For protist growth media, serum is often heat-inactivated at 56°C for 30 minutes to ensure the inactivation of blood components that may inhibit propagation.
Tryptose
Tryptose is an enzymatic digest of protein commonly used in microbial culture media to provide uniformity for the cultivation of fastidious organisms. It is commonly used in media formulations as a source of vitamins and amino acids.
Yeast extract
Yeast extract is prepared as a water-soluble extract of autolyzed Saccharomyces cerevisiae yeast cells. During autolysis, endogenous yeast digestive enzymes break down protein content into peptides and amino acids, which can be used by protists as a source of nitrogen. Additionally, yeast extract provides an essential source of water-soluble B-complex vitamins, carbohydrates, and free glutamic acid.8

Preservation
There are currently over 100 protocols used for the preservation of ATCC protozoan and algal strains. Below, we describe some general guidelines that can be applied in the preservation of these organisms. However, optimal protocols for preservation of a given strain should be empirically determined.
Presently, most strains of algae and protozoa maintained at ATCC are stabilized in the frozen state within liquid nitrogen freezers.9,10 In this configuration, most protozoa and algae can be stored indefinitely as cryopreserved cultures. Protist strains that make a desiccation-resistant cyst stage and many algal strains are amenable to storage in the lyophilized state.11,12 However, preservation by drying or lyophilization is less stable than freezing material and is not recommended for protist cultures.
There are many advantages of preservation that far outweigh the required investment in the equipment and supplements needed to maintain living cultures. These advantages include:
- Overall safety of stocks against loss due to equipment failure or contamination by other microbial organisms.
- Elimination of time, energy, and material costs associated with the maintenance of strains not currently in use.
- Insurance against phenotypic drift associated with prolonged passage, due to genetic instability and/ or selective pressures.
- Generation of a standard working stock that can be used for a series of experiments.
Cryopreservation
Overview
Generally, freezing a suspension of living cells may result in a decrease in viability.13,14 As the suspension is cooled below the freezing point, ice crystals begin to form and the concentration of solutes in the suspension increases. Additionally, the formation of intracellular ice crystals can result in the damage of cellular structures. This can be minimized if water within the cells is allowed to escape by osmosis during the cooling process. A slow cooling rate, generally -1°C to -10°C per minute, facilitates this progression. However, as the cells lose water, they shrink in size and will quickly lose viability if they surpass a minimum threshold volume. The addition of cryoprotectant agents, such as glycerol, methanol, or dimethylsulfoxide (DMSO), will mitigate these effects.15,16
The standard procedure for cryopreservation is to freeze materials slowly until they reach a temperature below -70°C in medium that contains a cryoprotectant. Vials are then transferred to a liquid-nitrogen freezer to maintain cultures at temperatures below -130°C. Successful recovery of cryopreserved protists requires the rapid thawing of the suspension in a 35°C water bath to avoid ice recrystallization as the frozen material warms. The entire contents of the vial are then transferred to an appropriate growth medium.
In general, there are numerous factors that can affect the viability of recovered protists. Among these, the rate of thawing is perhaps the most critical to successful recovery of cells from cryopreservation, which is why it must be done rapidly and carefully, particularly with sensitive types of protists. Slow hydration of the thawed sample with the growth medium (as described in Recovery Method 2 and Recovery Method 3 below) is one of the most effective techniques to counteract the deleterious effects of thawing.
Generally, it is recommended to harvest cultures that are in late log phase or early stationary phase growth, rather than during active growth. Larger cells that form food vacuoles are especially sensitive during cryopreservation and will generally exhibit greater survival rates when starved before preservation (starvation occurs in stationary phase).
Contact ATCC for more information on the cryopreservation of protozoa and algae.
Freeze medium
Glycerol, methanol, and DMSO are the most common cryopreservative agents used for protozoa and algae. DMSO is the most widely used cryoprotectant and can be used with most strains of protozoa. The concentration of DMSO used depends on the strain and can vary from 2.5% to 12.5%. For many algal strains, methanol is preferred as a cryoprotectant at a concentration ranging from 3% to 10%. Glycerol is the least effective cryoprotectant for protozoa and algae; however, certain strains, such as the Crypthecodinium, can only be frozen using this compound. Depending on the strain, glycerol is generally used at concentrations ranging from 10% to 28%.17 Examples of cryopreservatives used in frozen protist preparations are listed in Table 5.
Use only reagent-grade cryoprotectants that are stored in aliquots protected from light. ATCC offers DMSO (ATCC 4-X) that has been thoroughly tested for use. Overall, the optimum formulations for individual protists strains must be determined empirically.
Table 5. Examples of frozen protist preparations
| Genus | Cryoprotectant* |
|---|---|
| Acanthamoeba | 7.5% DMSO |
| Babesia | 10% Glycerol |
| Crypthecodinium | 7.5% Glycerol |
| Entamoeba | 10% DMSO + 3.5% sucrose (w/v) + 20% heat-inactivated bovine serum |
| Euglena | 3% - 5% Methanol |
| Giardia | 12% DMSO + 4% sucrose (w/v) |
| Hartmannella | 7.5% DMSO |
| Leishmania | 5% DMSO or 10% Glycerol |
| Ochromonas | 10% DMSO or 7.5% Methanol |
| Plasmodium | 28% Glycerol + 3% sorbitol (w/v) + 0.65% NaCl (w/v) or Glycerolyte 57 |
| Tetrahymena | 11% DMSO |
| Toxoplasma | 7.5% DMSO + 25% Heat-inactivated fetal bovine serum |
| Trichomonas | 5% DMSO |
| Trypanosoma | 5% DMSO |
*Dilute cryoprotectant in growth medium
Equipment
A. Cryopreservation vials
There are two materials to choose from for cryopreservation vials: glass or plastic. Glass vials are more difficult to work with, since they need to be sterilized before use, they need to be sealed with a hot flame, and they can be difficult to open. However, they are preferred for long-term storage (many years) of valuable cultures and are considered fail-safe once properly sealed.
If cryopreservation in glass ampoules is not possible, plastic vials can be used. Plastic vials come in two varieties: those with an internal thread and silicone gasket, and those with an external thread. Vials with an internal-thread were the first commercially available, but have some disadvantages over the external-thread version. For example, while the silicone gasket provides an excellent seal, it needs to be tightened just right; the vial will leak if the seal is too tight or too loose. ATCC cultures are currently preserved in plastic vials.
B. Controlled-rate freezing chambers
There are several means to achieve a controlled cooling rate. The best method involves the use of a computer-controlled, programmable electronic freezing unit (such as Thermo Scientific CryoMed Freezers), which rigorously maintains this rate of cooling through Recovery Methods latent heat of fusion. Such equipment is relatively expensive and only absolutely necessary for the most sensitive strains.
A less costly approach is to place the cryopreservation vials into an insulated chamber and cool in a mechanical freezer at -70°C or colder until the culture reaches -40°C or colder; cultures are then transferred to liquid nitrogen storage. ATCC offers a safe and effective freezing chamber which achieves a cooling rate very close to -1°C per minute (CoolCell LX Alcohol-Free Cryopreservation Container). Alternatively, the vials can be placed into a polystyrene box, with 15-mm (3/4 inch) thick walls and 1L capacity that is packed with paper, cotton wool, or foam peanuts for insulation.
Liquid nitrogen freezing storage

The ultra-low temperatures (below -130°C) required for long-term storage can be maintained by specialized electric freezers, or more commonly, by liquid nitrogen freezers. There are two basic types of liquid nitrogen storage systems: Immersing vials in the liquid or holding vials in the vapor phase above the liquid. The liquid-phase system holds more nitrogen and thus requires less maintenance. However, there is always a chance that some liquid will enter improperly sealed vials, which may then explode when retrieved. For this reason, ATCC strongly recommends storage in the vapor-phase.
Vapor-phase storage systems create a vertical temperature gradient within the container. The temperature in the liquid nitrogen at the bottom will be -196°C, whereas the temperature at the top will vary depending upon the amount of liquid nitrogen at the bottom and the length of time the container is opened. To ensure the safe storage of cells, maintain sufficient levels of liquid nitrogen in the container so that the temperature at the top is -130°C or colder. All storage systems should be equipped with temperature alarms.
Cryopreservation procedure
The procedure described below offers general guidelines that can be applied in the preservation of protist strains. ATCC recommends that the optimal cryopreservation procedure be determined empirically for each species.
1. Propagate and harvest protist cultures
Incubate the protist strain under the recommended conditions and harvest in vitro strains in late logarithmic to early stationary phase of growth. There are six basic harvesting protocols, some being more effective than others depending on the motility of the protist and its sensitivity to centrifugation:
- Harvest method 1 – Simply allow the cells to settle to the bottom of a culture vessel and remove the overlaying culture fluid with a pipette.
- Harvest method 2 – Some cells can be gently concentrated using gravity filtration through a nylon mesh screen which does not allow cell passage.
- Harvest method 3 – For strains which attach to the wall of the culture vessel, detach the cells by icing the culture for 10 minutes, gently invert the culture vessel several times to mix, and remove the culture fluid with a pipette.
- Harvest method 4 – For strains cultured on agar media, add broth medium directly to the agar, and suspend cells by rubbing the agar surface with a spread bar.
- Harvest method 5 – If centrifugation is required, use the lowest speed that will allow pelleting of the cells. For sensitive cell types, following resuspension transfer to a tissue culture flask and allow the sample to remain undisturbed for 1 hour prior to the addition of the cryoprotectant. This will allow cells damaged by the harvesting procedure to undergo some repair.
- Harvest method 6 – Vertebrate blood parasites propagated in host animals can be harvested when the parasitemia has reached the desired level. The blood is withdrawn from the host animal using a syringe containing a small amount of anticoagulant.
2. Add the cryoprotective solution
After harvesting the culture, add the appropriate cryopreservative agent to the protist culture. ATCC recommends that cryopreservative solutions should be prepared as a 2× concentrated stock and mixed at a ratio of 1:1 with the cell suspension. For sensitive organisms, the concentrated cryoprotective solution should be added in three equal aliquots at 2-minute intervals. Dispense 0.5 to 1.0 mL of the final suspension into a sterile cryovial.
3. Equilibrate the cultures
Allow the cell suspension to equilibrate with the cryoprotective solution prior to cooling. This equilibration time should be at least 10 minutes, but no longer than 30 to 60 minutes. Exposing the cells to the cryoprotectant for too long may be detrimental to cells. Tolerance to DMSO will vary with the organism and should be determined prior to freezing. Please view the product sheet for additional information regarding culture equilibration.
4. Cool the sample and store
Algae and protozoa are generally frozen using a controlled cooling rate. There are three controlled cooling rates routinely used. In some cases an uncontrolled cooling rate can yield sufficiently high recovery and may be essential for the recovery of some strains. Overall, frozen specimens of protozoa and algae must be maintained at temperatures below -130°C for optimum stability. Contact ATCC Product Experience for the appropriate cooling parameter of your culture of choice.
- Controlled cooling 1 – This cooling cycle is effective for many strains. Cool the suspension at a rate of 1°C per minute from room temperature to -40°C. The ampoules are plunged into liquid nitrogen from -40°C and stored in liquid nitrogen vapor. Supercooling of the cultures is often encountered when using this cooling program, which can be a problem for certain sensitive strains.
- Controlled cooling 2 – Some protozoa are sensitive to supercooling and therefore a triphasic cooling program was developed to deal with these strains. The triphasic cooling program is as follows: 10°C per minute from room temperature to the heat of fusion, and 1°C per minute from the heat of fusion to -40°C. The ampoules are then plunged into liquid nitrogen and stored in liquid nitrogen vapor. The one disadvantage of this cooling cycle is that certain strains may be sensitive to cold shock.
- Controlled cooling 3 – In order to minimize cold shock but avoid supercooling, the following compromise cooling cycle has been developed: 1°C per minute from room temperature to 4°C; 10°C per minute from 4°C to the heat of fusion; 1°C per minute from the heat of fusion to -40°C. The ampoules are then plunged into liquid nitrogen.
- Uncontrolled cooling 1 – Some strains can be cryopreserved using an uncontrolled cooling rate. The ampoules are placed at the bottom of a mechanical freezer at -55°C to -60°C for one hour and then transferred to liquid nitrogen vapor.
- Uncontrolled cooling 2 – Some intracellular blood parasites must be frozen using an uncontrolled cooling cycle. When the parasitemia has reached the desired level, the blood is withdrawn and combined with an equal amount of a 2× concentrated cryoprotective solution. After equilibration, plunge the ampoules directly into a dry ice/ethanol bath for 2 minutes. Immediately transfer the sample to liquid nitrogen vapor.
5. Sample recovery
To ensure the viability of a protist strain upon recovery, strains should be thawed rapidly and carefully. There are three methods that can be used to recover frozen samples. Contact Product Experience for the recommended recovery method of your culture of choice.
- Recovery method 1 – Transfer the frozen cultures directly from liquid nitrogen to a water bath set at 35°C. Immerse the ampoule just enough to cover the frozen material, do not agitate the sample. For most strains, the thawed contents can be added directly to a standard culture vessel. For some strains which are sensitive to osmotic shock, increasing the volume of the frozen suspension from mL to 1.0 mL can be beneficial. As the volume of the frozen suspension is increased, the time for contents of the ampoule to completely thaw will be increased, and the cells will have more time to adjust osmotically.
- Recovery method 2 – Recovery of some sensitive strains can be enhanced by increasing the osmolarity of the medium used for initial establishment of the culture. For some strains of Tetrahymena, Euplotes, and Glaucoma, this may be critical to survival. The following protocol can be used to thaw these sensitive organisms:
- Remove the frozen ampoule from liquid nitrogen and aseptically add 0.5 mL of medium containing 8% sucrose (w/v) (See: NOTE 11).
- Screw the cap on tightly and place in a 35°C water bath.
- Transfer the thawed contents to medium with 4% sucrose (w/v).
- Incubate in the normal fashion for Tetrahymena but transfer to medium without sucrose when the culture is in the stationary phase.
- For bacterized cultures on the following day, pick viable cells with a drawn-out Pasteur pipette and transfer to fresh medium without sucrose.
- Recovery method 3 – Neither method 1 nor method 2 is suitable for sensitive marine organisms and some freshwater organisms. In these cases, the following protocol should be followed:
- Transfer the frozen cultures directly from liquid nitrogen to a water bath set at 35°C. Immerse the ampoule just enough to cover the frozen material, do not agitate the sample.
- Gently remove contents with a Pasteur pipette and expel slowly into a T-25 tissue culture flask. Place the flask in a horizontal position and incubate at room temperature.
- At 15-minute intervals, add 0.25 mL of the appropriate medium drop-wise. Continue until the final volume is 2.0 mL. After the final aliquot is added, allow the flask to remain undisturbed for 15 minutes.
- Add 0.5 mL of the appropriate medium drop-wise at 15-minute intervals until the final volume is 4.0 mL. Allow the flask to remain undisturbed overnight.
- In the morning of day 2, slowly add 4.0 mL of the appropriate fresh medium. Allow the sample to remain undisturbed overnight.
- In the morning of day three proceed as follows:
- If the culture has free-swimming cells or unattached nonmotile cells, gently agitate the flask and transfer 1 mL to a fresh T-25 flask containing 10 mL of the appropriate medium. Incubate thereafter using normal protocol. Maintain both cultures in parallel.
- If the culture has attached cells, gently remove all but approximately 1 mL of the culture fluid with a pipette and transfer this material to a fresh T-25 flask. Add 10 mL of the appropriate medium to the decanted flask. Maintain both cultures in parallel.
Note 11:
Manipulations of the frozen ampoule before placing in the water bath should be done as quickly as possible to avoid warming the contents at a suboptimal rate, which can damage the cells.
Special Hazards
Care must be taken when preserving protists. Problems such as contamination, breakage of glass ampoules during handling and storage, dispersal of materials when opening glass ampoules, and the handling of liquid nitrogen must all be considered. To prevent contamination and the dispersal of material, aseptic technique must be followed. This can include the decontamination of all equipment and vials as well as performing all preparations in a biological safety cabinet. Additionally, protective clothing should be worn during preparation to prevent contamination as well as to guard against harm due to contact with liquid nitrogen.

Biosafety and disposal
Biosafety
The need for precautions when experimenting with protist cultures depends upon the source and nature of the biological material, the experimental procedure, and the laboratory/containment conditions. Since every situation is different, the risks need to be identified for each individual strain and the appropriate precautions need to be taken before any work begins.
More information on risk assessment and precautions can be found in the CDC publication Biosafety in Microbiological and Biomedical Laboratories.18 The text of this publication is available in its entirety online (https://www.cdc.gov/labs/bmbl/index.html).
ATCC assigns a biosafety level (BSL) to each strain for the purposes of packaging for safe shipment. ATCC follows federal biosafety guidelines and takes several factors into consideration when assessing a potential hazard, and in some cases the ATCC assigned biosafety level is more restrictive. Generally, ATCC only ships and stores protist strains with a biosafety level assignment of 1, 2, or 3.
Biosafety level 1
- Work involving well-characterized strains not known to consistently cause disease in immunocompetent adult humans.
- Work can be conducted on the bench top using aseptic technique, no special containment equipment or facility is required.
Biosafety level 2
- Work involving strains that pose a moderate hazard to healthy adult humans.
- Work should be conducted in designated biological safety cabinets within laboratories with restricted access.
Biosafety level 3
- Work involving indigenous or exotic agents that may cause serious or potentially lethal disease via inhalation.
- Work should be conducted in biological safety cabinets localized inside a specialized BSL-3 containment facility within laboratories with restricted access.
As the recipient of a protist, take into account not only the nature of the material but also the manipulations employed during its handling when assessing the potential laboratory risk. Keep in mind that there will be situations where the intended use of an agent may require more stringent precautions than associated with the assigned biosafety level.18
Disposal of infectious materials
All cultures, stocks, and potentially infectious materials need to be properly decontaminated prior to disposal. The written method for proper decontamination should be available in the laboratory and BSL facility. Several methods of sterilization include chemical disinfection, incineration, use of an autoclave, or any other validated decontamination method. More information on the disposal of protist cultures can be found in the Center for Disease Control (CDC) publication Biosafety in Microbiological and Biomedical Laboratories.18
Protist authentication
Phenotypic characterization
Morphological data is an essential part of authenticating protist strains as it provides preliminary information regarding the identity, health, and purity of each strain. In most cases, this information can be obtained from living or stained material using light microscopy. For algal species, morphological traits analyzed include colony formation, unicellular morphology, motility, and the presence or absence of filamentous, siphonous, or parenchymatous structures. For protozoan species, many strains are grouped by their method of locomotion. Amoeboids, such as the Amoeba, are characterized by transient pseudopodia. Ciliates, such as the Paramecium, are characterized by their multiple, short cilia. Finally, flagellates, such as the trypanosomatids are distinguished by having only a single flagellum which is closely associated at its base with the kinetoplast.
In addition to the standard morphological assessment by microscopy, determination of culture purity should be performed using sterility testing and/or mycoplasma testing where applicable. Depending on the requirements of the protist strain, a xenic culture may contain bacteria or other microorganisms required as a food source. Further, some amoeboid protists harbor intracellular pathogens; an example of this includes the amoeba Hartmannella vermiformis, which is often host to the bacterium Legionella pneumophila.
Genotypic characterization
Due to the variety of protist taxa, molecular authentication is essential in ensuring strain identity. Sequencing of the nuclear 18S rRNA gene and the associated internally transcribed spacer (ITS) regions have been shown to be useful tools for analyzing protists at the species level. However, for some protist species, the 18S rRNA/ITS sequences may not allow adequate species determination. In this instance, DNA barcoding is used. DNA barcoding is a taxonomic method that analyzes a short genetic marker in the DNA of a strain to confirm its species. The genetic marker examined is dependent on the organism; for example, cytochrome c oxidase (cox1) has been demonstrated to effectively separate different species of Paramecium.19

General representation of eukaryotic 18S, 5.8S, and 28S rRNAs. Genes are flanked by the external transcribed spacers (5' and 3' ETS) and separated by the internal transcribed spacers (ITS1 and ITS2).

Protist applications
Bioassays
Several species of protists have been used in bioassay procedures for the detection and measurement of very small quantities of vitamins and growth factors; axenic cultures using high purity defined media are essential for this work. Examples of protists that require specific vitamins for growth and that have been used in these assays include Euglena gracilis and Ochromonas sp. These organisms can be used in estimation of vitamin B12 at concentrations down to 1 ng/mL.19
Biofuel production
Recently, efforts have been made to generate sustainable energy via the production of biofuels from commercially available products such as corn and sugar cane. However, these novel sources of energy require food to be used as fuel rather than for human sustenance. To resolve these issues, current biofuel research is focusing on the use of algae as an alternative to fossil fuel. There are many advantages to using algae as a source of biofuel. Like fossil fuels, algae releases carbon dioxide when burned. Further, algae are a renewable resource that can survive on land that is not suitable for crop growth. Furthermore, the lipid portion of an algal biomass can be extracted and converted to biodiesel. Lastly, the carbohydrate content of an algal biomass can be fermented into bioethanol and biobutanol. Overall, the use of algae in bioenergy would create a sustainable form of energy without diminishing the food supply.
Fatty acid production
Docosahexaenoic acid (DHA) is an omega-3 fatty acid that is a primary structural component of the brain and retina. This fatty acid is produced by a number of sources including fish and the protist Crypthecodinium cohnii. C. cohnii, a free-living heterotrophic dinoflagellate, is an excellent source of DHA and is actively used by food manufacturers to provide DHA as a food supplement to reduce the risk of stroke in human adults.20 Further, this fatty acid is often supplemented in infant formula to promote proper eye care and brain development in babies and young children. Other protist genera that are significant sources of DHA include Aurantiochytrium, Thraustochytrium, and Schizochytrium.
Diagnostics
Systemic lupus erythematosus (SLE) is a serious autoimmune disorder that is difficult to diagnose, but can be identified in some patients by the presence of autoantibodies directed against native double-stranded DNA (nDNA). Among the battery of tests used to diagnose SLE, is the autoantibody test based on a Crithidia luciliae substrate.21 C. luciliae, a unicellular insect trypanosome, contains an intracellular organelle, termed the kinetoplast, which contains a high concentration of nDNA. This structure is used as the primary target for the Crithidia luciliae indirect immunofluorescence test (CL-IFT) as existing SLE autoantibodies will bind to the nDNA within the C. luciliae kinetoplast. To perform the CL-IFT assay, C. luciliae kinetoplasts are bound to a glass slide and are allowed to interact with a patient’s serum sample. Through the use of fluorescent-bound anti-human antibodies, the presence of bound SLE autoantibodies can be detected by fluorescent microscopy.
In recent years, molecular techniques have become more commonplace in the diagnosis of parasitic infections. To support this research, ATCC holdings include approximately 1000 strains of parasitic protozoa representing a variety of organisms including Toxoplasma gondii, Giardia lamblia, and species within the genera Plasmodium, Babesia, Trypanosoma, Leishmania, Trichomonas, and Entamoeba.
Consumable products
Algae strains are used in a number of consumable products. Chlorella have been used as a food supplement due to their high abundance of proteins, lipids, and vitamins. Large scale cultivation is attainable in artificial ponds. Green algae are often used in traditional Asian cuisines because of their nutritional value; an example of this is the use of algae wraps in sushi. Additionally, sulfated polysaccharides known as carrageenan are extracted from red algae and used as thickening agents in various dairy products.22 Products containing carrageenan include sour cream, mayonnaise, cheese, and yogurt.
Download a PDF of our protistology culture guide
Get the guide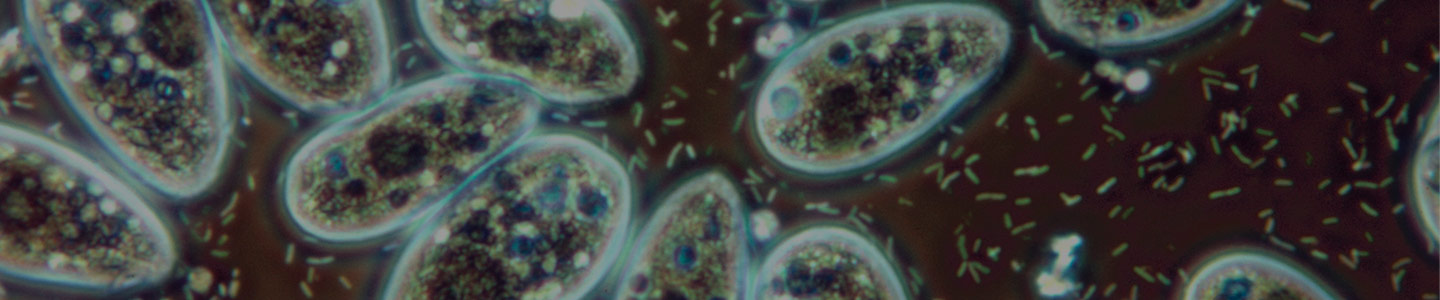
Glossary
Algae. Photosynthetic protists. This group does not include the blue-green algae, which are considered bacteria.
Amoeboids. Single celled organisms characterized by an irregular shape and locomotion via pseudopodia.
Autotroph. An organism that functions as a primary producer in the food chain. These organisms obtain energy and nutrients through photosynthesis.
Axenic. A culture containing a population of only a single organism.
Bacterized Culture. A culture that contains bacteria to be used as a food source for the protist.
Cilia. Hair-like organelles that are typically short and present in large numbers across the surface of a cell. These organelles are required for cellular locomotion.
Ciliates. Protozoans that require cilia for locomotion.
Clonal Monoprotist. A monoprotist derived from a single cell that has undergone repeated cloning. This culture may contain an uncharacterized bacterial community but no other eukaryotes.
Cyst. The resting, dormant, or resistant stage in the full life cycle of many protozoan species. This form may play a role in dispersal of the organism.
Diclonal Diprotist. A culture in which both protist species are clonal and have undergone repeated cloning. This culture may contain an uncharacterized bacterial community but no other eukaryotes.
Diprotist. A culture that consists of only two protist species and contains bacteria, but no other eukaryotes. This is the typical situation for most heterotrophic protists that require another protist species as a food source.
Filamentous Algae. Algae that undergo cell division but remain connected, forming long filaments of attached cells. Each cell within the filamentous alga has its own internal structures and is capable of photosynthesis.
Flagellates. Protozoa that have one or more whip-like flagella for locomotion.
Heterotroph. An organism that consumes other organisms in the food chain. These organisms must rely on an organic source of carbon that originated from another living organism.
Latent Heat of Fusion. Heat energy that is released when ice crystals begin to form.
Mixotroph. An organism that is capable of existing as either an autotroph or heterotroph.
Monoclonal Diprotist. A clonal culture of a protist species co-existing with one other non-clonal protist species that usually serves as its food source. This culture may contain an uncharacterized bacterial community but no other eukaryotes.
Monoprotist. A culture that contains a single protist species and bacteria, but no other eukaryotes.
Monoxenic. A culture containing a population of only two organisms, the organism being actively studied and the food source. See monoprotist.
Osmotrophy. A heterotrophic organism that absorbs dissolved nutrients by pinocytosis.
Parasitemia. The quantitative content of parasites in the blood. Parasitemia is expressed as a percentage and is equal to the number of infected red blood cells (RBC) relative to the total number of RBC counted (ie, if 1 out of every 5 cells counted is infected, parasitemia = 20%).
Parenchymatous Algae. Algae that comprises boxy, plant-like cells with a complex multicellular structure.
Phagocytosis. The process of engulfing and ingesting particles through the extension of pseudopodia.
Phagotroph. A heterotrophic organism that ingests particulates and uses intracellular digestion through phagocytosis.
Pinocytosis. The ingestion of fluid into a cell by engulfing and internalizing the fluid in portions of the cell membrane that pinches off into vesicles.
Protozoa. Heterotrophic protists that are primarily phagotrophic.
Pseudopodia. Temporary projections of eukaryotic cells that extend and contract to allow cellular movement.
Siphonous Algae. Algae with a morphology resembling filamentous algae, but consisting of only a single cell with branching sections.
Sporozoa. Parasitic protozoa that are typically immobile.
Supercooling. The process of lowering the temperature of a substance below its freezing point without it becoming a solid.
Xenic. A culture medium containing one or more organisms.
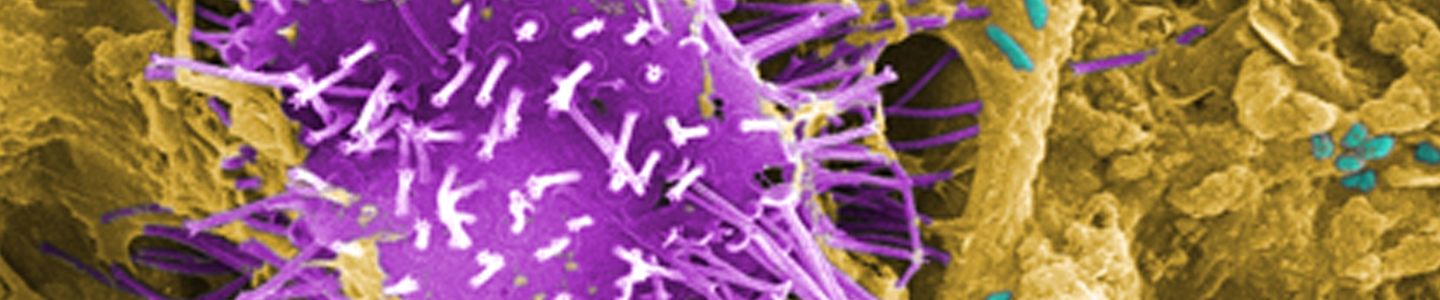
References
- Adl, S.M. et al. The new higher level classification of eukaryotes with emphasis on the taxonomy of protists. J Eukaryot Microbiol 52, 399-451 (2005).
- Adl, S.M. et al. The revised classification of eukaryotes. J Eukaryot Microbiol 59, 429-93.
- Simpson, A.G. Cytoskeletal organization, phylogenetic affinities and systematics in the contentious taxon Excavata (Eukaryota). Int J Syst Evol Microbiol 53, 1759-77 (2003).
- Moreira, D. et al. Global eukaryote phylogeny: Combined small- and large-subunit ribosomal DNA trees support monophyly of Rhizaria, Retaria and Excavata. Mol Phylogenet Evol 44, 255-66 (2007).
- Cavalier-Smith, T. & Choao, E.E. Phylogeny of Choanozoa, Apusozoa, and other Protozoa and early eukaryote megaevolution. J. Mol. Evol. 56, 540-563 (2003).
- Bakkeren, G., Kamper, J. & Schirawski, J. Sex in smut fungi: Structure, function and evolution of mating-type complexes. Fungal Genet Biol 45 Suppl 1, S15-21 (2008).
- Convention, T.U.S.P. The United States Pharmacopeia 31- The national formulary 26 (The United States Pharmacopeial Convention, Inc., Rockville, MD, 2008).
- Diagnostics, B. Difco & BBL Manual, Manual of Microbiological Culture media (Copyright by Becton Dickinson and Company, Sparks, MD, 2009).
- Diamond, L.S. Freeze-preservation of protozoa. Cryobiology 1, 95-102 (1964).
- Hwang, S., Davis, E.E. & Alexander, M.T. Freezing and viability of Tetrahymena pyriformis in dimethylsulfoxide. Science 144, 64-65 (1964).
- McGrath, M.S. & Daggett, P.M. Cryopreservation of flagellar mutants of Chlamydomonas reinhardtii. Can. J. Bot. 55, 1794-1796 (1977).
- McGrath, M.S., Daggett, P.M. & Dilworth, S. Freeze-drying of algae: Chlorophyta and Chrysophyta. J. Phycol. 14, 521-528 (1978).
- Mazur, P. The role of intracellular freezing in the death of cells cooled at supraoptimal rates. Cryobiology 14, 251-72 (1977).
- Mazur, P. Cryobiology: the freezing of biological systems. Science 168, 939-49 (1970).
- Fahy, G.M. The relevance of cryoprotectant "toxicity" to cryobiology. Cryobiology 23, 1-13 (1986).
- Meryman, H.T. Cryoprotective agents. Cryobiology 8, 173-83 (1971).
- ATCC. ATCC Preservation Methods: Freezing and Freeze-drying (eds. Simione, F.P. & Brown, E.M.) (American Type Culture Collection, 1991).
- United States Department of Health and Human Serves, C.f.D.C., and National Institutes of Health. Biosafety in Microbiological and Biomedical Laboratories (U.S. Government Printing Office, HHS Publication No. (CDC) 21-1112, Washington DC, 2009).
- Barth, D., Krenek, S., Fokin, S.I. & Berendonk, T.U. Intraspecific genetic variation in Paramecium revealed by mitochondrial cytochrome C oxidase I sequences. J Eukaryot Microbiol 53, 20-5 (2006).
- Beach, D.H., Harrington, G.W., Gellerman, J.L., Schlenk, H. & Holz, G.G.J. Biosynthesis of Oleic acid and docosahexaenoic acid by a heterotrophic marine dinoflagellate Crypthecodinium cohnii. Biochim Biophys Acta 136, 16-24 (1974).
- Slater, N.G.P., Cameron, J.S. & Lessof, M.H. The Crithidia luciliae kinetoplast immunofluorescence test in systematic lupus erythematosus. Clin. Exp. Immunol. 25, 480-486 (1976).
- Flett, K.L., Duizer, L.M. & Goff, H.D. Perceived creaminess and viscosity of aggregated particles of casein micelles and kappa-carrageenan. J Food Sci 75, S255-62.