Authors: Elizabeth Turner-Gillies, PhD; Brian Shapiro, PhD; and Fang Tian, PhD
Introduction
Results and Discussion
Conclusion
References
Abstract
Antiviral vaccines are essential for preventing epidemic disease; however, the production of these therapeutics is often limited by low-yielding manufacturing processes. Similarly, the development of gene therapies is constrained during the large-scale production of viral vectors, such as adeno-associated virus (AAV) delivery platforms for gene transfer.
To speed the pace of these various areas of bioproduction, ATCC addressed the need for efficient viral replication by optimizing three cell lines commonly used in virus manufacturing. Here, we used CRISPR/Cas9 gene-editing technology to develop STAT1 and BAX knockout cell lines capable of producing high-titer viral stocks. These newly created cell lines can produce model clinical viruses and adeno-associated viruses at titers much higher than the parental cell lines, providing an efficient method for biopharmaceutical companies to increase production while reducing associated costs.
Download a PDF of this application note
Download NowIntroduction
The manufacture of antiviral vaccines using mammalian cell systems requires the production of large quantities of viral particles in a scaled-up tissue culture system. A handful of historical cell lines like Vero (African green monkey kidney) and MDCK (canine kidney) are approved by the FDA for the production of many viral vaccines. However, other than clone selection and cell culture media adaption, relatively little has been done to improve the inherent viral production capacity of these cell lines. Additionally, there has been a resurgence in gene therapy, driven by the development of safe and easy-to-use AAV vectors. As in the process of manufacturing vaccines against viral infections, a bottleneck in the production of AAV-derived gene therapy products is at the point of infection, replication, and egress of the virus in the HEK 293 cell line.
The emergence of new, pathogenic viral strains and the demand for therapies for genetic diseases contribute to the urgent need to accelerate manufacturing processes. While there are many points at which improvements could be made to speed virus production, optimizing the cell lines used in the process has the potential to yield the most benefit with regards to reduction in cost and time. Some strategies that could be used to optimize virus-producing cell lines include optimizing virus-producing cells (VPCs) for viral vector uptake, adjusting the VPC’s growth mode from adherent to suspension or high-density culture, and genetically engineering VPCs to enhance their viral production yield.
Of the above mentioned areas of improvement, ATCC took a step to create high-titer viral production cell lines through CRISPR/Cas 9 gene editing. With the design strategy of enhancing viral particle yield by eliminating the interferon response pathways and improving the VPC’s survival via deleting/down regulating of pro-apoptotic genes, we employed two approaches. The first was to exploit the fact that cells rely on interferon-elicited pathways as defense against viral infections. A major effector of interferon signaling is via the STAT1 protein. Phosphorylation and the resulting self-dimerization of STAT1 induces this intracellular signaling protein to translocate to the nucleus, resulting in a number of antiviral, antiproliferative, and immunoregulatory responses by the cell. Thus, ablating STAT1 from this system results in a profound reduction in the cell’s antiviral response and a concomitant increase in viral yield.¹,² In this regard, ATCC scientists used CRISPR/Cas9 genome-editing technology to knock out the STAT1 gene in Vero (ATCC CCL-81) and MDCK (ATCC CCL-34) cells as a strategy to enhance virus production. With the STAT1 gene disrupted, the STAT1 protein is not synthesized and thus the interferon signaling cascade is disrupted. Without the cell’s defense against the viral infection, we hypothesized that virus production would be significantly enhanced in the newly created Vero.STAT1 KO (ATCC CCL-81-VHG) and MDCK.STAT1 KO (ATCC CCL-34-VHG) cell lines as compared to that of the parental cell lines.
The second approach that was taken involved knocking out both the BAX and STAT1 genes. BAX protein is a central activator of apoptosis via the intrinsic pathway and has functions in virally induced cell death. Indeed, BAX has been identified by Cuconati and colleagues in 2002 to limit AAV production via the apoptotic response.³ Thus, we hypothesized that deletion of the BAX gene product from VPCs would increase cell survival and remove another hurdle to enhanced virus production. As with the abovementioned modified cell lines, we used CRISPR/Cas9 genome-editing technology to knock out both BAX and STAT1 from the AAV-producing parental HEK 293 cell line (ATCC CRL-1573). As in the other models, without the VPC’s antiviral defense and major apoptotic regulator, we anticipated that AAV production would be enhanced in the newly generated 293.STAT1 BAX KO (ATCC CRL-1573-VHG) as compared to that of the parental cell line.
Overall, we provide evidence indicating that the ATCC STAT1 and STAT1/BAX knockout cell lines produce model clinical viruses at titers 10-fold higher than the corresponding parental cell lines; these gene-edited cell lines have the potential to significantly reduce costs and time expenditures that are associated with virus and viral vaccine production. Thus, these optimized cell lines are not only good candidates for enhancing virus titers for vaccine development, they also provide useful tools to package lentiviral, adenoviral, and retroviral vectors for the delivery of DNA sequences into target cells for various applications such as gene therapy and cancer immunotherapy.
Results and discussion
STAT1 and STAT1/BAX knockout strategy and execution
We employed the CRISPR/Cas9 genome-editing platform to generate the desired, targeted STAT1 knockout (Vero.STAT1 KO and MDCK.STAT1 KO) or STAT1 and BAX double knockout (293.STAT1 BAX KO) VPCs. Our strategy was to generate out-of-frame insertions and deletions near the start of STAT1 and BAX genes, which would result in a functional protein knockout. Because of the transient nature of the Cas9 construct transfection strategy, the constructs themselves were not integrated into the gene-edited clones. To execute on this strategy, single guide RNAs (sgRNAs) were designed and built to guide Cas9 to bind and cut the desired intronic regions in the STAT1 and BAX gene targets.
We then transiently transfected Cas9 and either the STAT1 gRNA or STAT1 and BAX gRNA constructs into the viral production cells. Once expressed within the cells, CRISPR/Cas9 created a double-strand break (DSB) in the appropriate gene. The cells’ non-homologous end joining (NHEJ) repair mechanisms then created small insertions and deletions at the repair site. After gene editing was complete, the clones were subjected to a battery of tests. Sanger sequencing was used to confirm that the correct frame-shift mutation occurred, and western blot was used to confirm that expression of the target genes was absent (Figure 1). Additionally, the VPCs were evaluated for off-target editing, stability, morphology, and cell growth. We found that the targeted gene knockout was very stable, and there was no off-target editing in similar sequences throughout the whole genome. Additionally, the VPCs displayed very similar morphology and equivalent growth rate as their parental cell lines.
Biofunctional evaluation of VPCs: Viruses for vaccine development
Once the Vero.STAT1 KO, MDCK.STAT1 KO, and 293.STAT1 BAX KO cell lines were created and quality control analyses were performed, we evaluated their virus production capacity as compared to that of their respective parental cell lines. Generally, the strategy for evaluating virus production was the same for all of the cell lines that we tested.
Vero.STAT1 KO and Vero cells were infected with Dengue virus type 2 strain New Guinea C (ATCC VR-1584); after seven days post-infection, the supernatants were harvested, serially diluted, and used to re-infect fresh cell cultures. Viruses within the cells were then subjected to immunofluorescence staining. High-content microscopy was used to capture fluorescence images of each well for the evaluation of infectious viral titer by TCID50 (Figure 2A and Figure 2B). When compared to the virus production capacity of Vero cells, a 10-fold increase in Dengue virus–infected cells was observed in the Vero.STAT1 KO cells. Additionally, RT-PCR was performed in the STAT1 KO and parental cell lines (Figure 2C), where we saw a 30-fold increase in Dengue viral genomes in the KO cell line.
We performed similar experiments in MDCK.STAT1 KO and the MDCK parental cell line. In this set of experiments, we infected both cell lines with Influenza A virus (H1N1; ATCC VR-1736) and harvested supernatants 48 hours post-transduction. Serial dilutions of the viral supernatants were again used to infect MDCK.STAT1 KO and MDCK cells, and high-content imaging and RT-PCR was performed as before (Figures 3A-C). Similar to the gene-edited Vero cells, a 10-fold increase in Influenza A virus–positive cells and a 2-fold increase in viral genomes was observed in KO cell line as compared to the parental MDCK cells.
Finally, to examine the 293.STAT1 BAX KO cell line’s virus-producing competence, we infected both the knockout and wild type HEK 293 cell lines with GFP-expressing Sendai virus (ATCC VR-105) and harvested supernatants 48 hours post-transduction. As before, serial dilutions of the viral supernatants were used to infect the knockout and wild type cell lines, and high-content imaging and RT-PCR was performed (Figures 4A-C). In line with the STAT1 KO cell lines, we saw a more than 5-fold increase in Sendai virus–positive cells in the KO cell line as compared to the parental HEK 293 cells. To examine the 293.STAT1 BAX KO cell line’s capacity of producing AAV, we transfected the cells with an AAV5 viral vector (ATCC VR-1523) and collected supernatants after 48 hours. Droplet digital PCR quantification of AAV5 viral genomes at 72 hours post-infection demonstrated an increase of about 80% when comparing 293.STAT1 BAX KO cells to the parental HEK 293 cells.
Conclusion
An increasing number of viral vaccines are manufactured in large-scale tissue culture systems using historical cell lines that are approved for vaccine manufacturing. In this study, we used CRISPR/Cas9 gene-editing techniques to increase the viral production efficiency of some of these historical cell lines.
Vero and MDCK cells were modified by CRISPR/Cas9 to produce a truncated STAT1 gene product that is quickly proteolyzed by the cell. Similarly, HEK 293 cells were genome edited to knock out expression of STAT1 and BAX. All three cell lines displayed increased viral production capacity. These highly validated cell lines were thoroughly characterized for viral production phenotype, stability of the STAT1 and STAT1/BAX double knockout genotype, and absence of off-target mutations.
ATCC’s cell lines for enhanced viral production have broad applicability for bioproduction in basic research and the pharmaceutical industry. Because of their ability to yield higher virus titers than their wild-type counterparts, these enhanced virus-producing cell lines have the potential to significantly reduce the costs associated with generating viral vaccines and gene therapies.

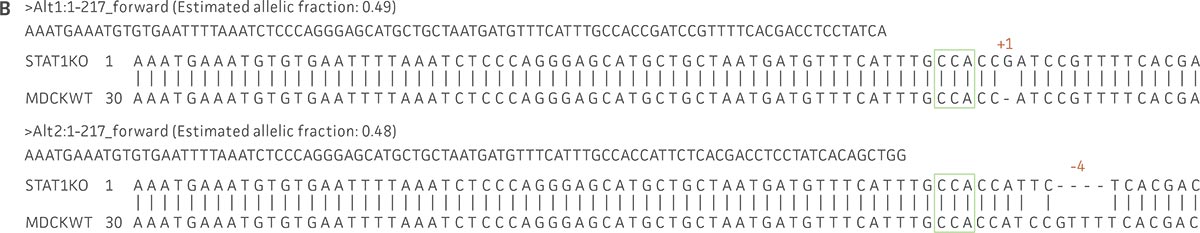
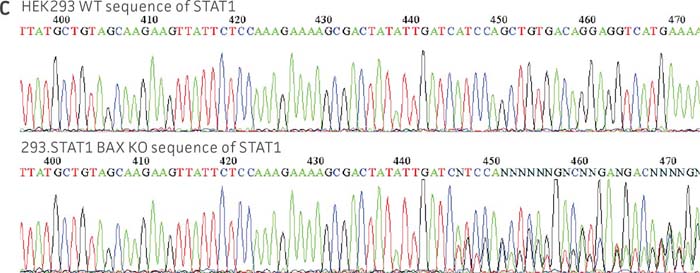
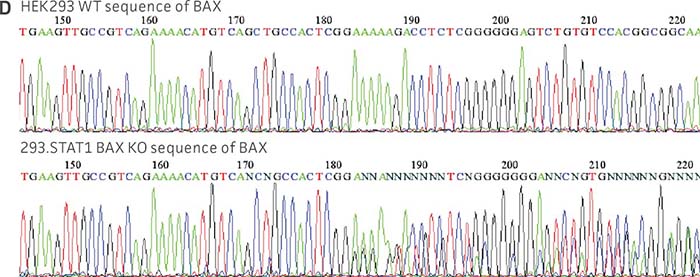
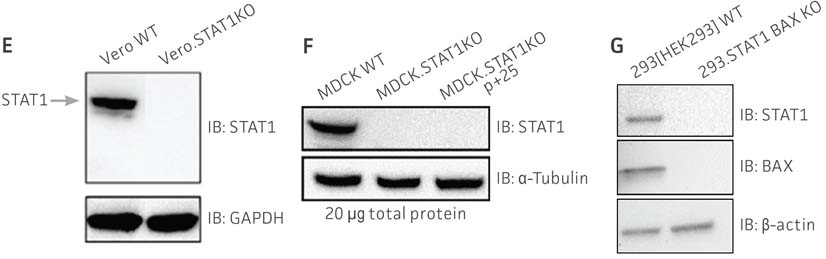
Figure 1: Molecular characterization of three VHG cell lines. (Figure A, Figure B, Figure C) Sanger sequencing of the STAT1 gene of all three virus-producing KO cells confirms the disruption of the STAT1 gene. (Figure D) Sanger sequencing of the BAX gene of 293.STAT1 BAX KO cells confirms BAX gene disruption. (Figure E-G) STAT1 and BAX immunoblot of total cellular protein from parental wild type and knockout cells confirms absence of both STAT1 protein or STAT1 and BAX protein expression in the enhanced virus-producing cell lines.

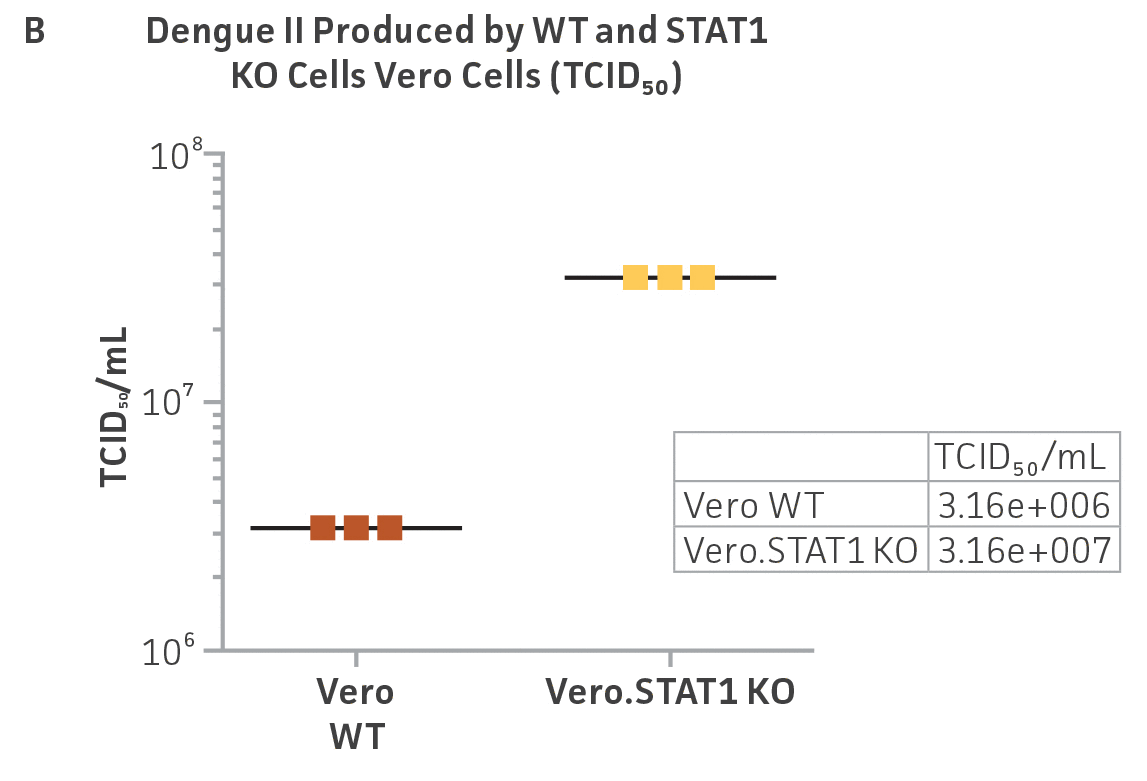
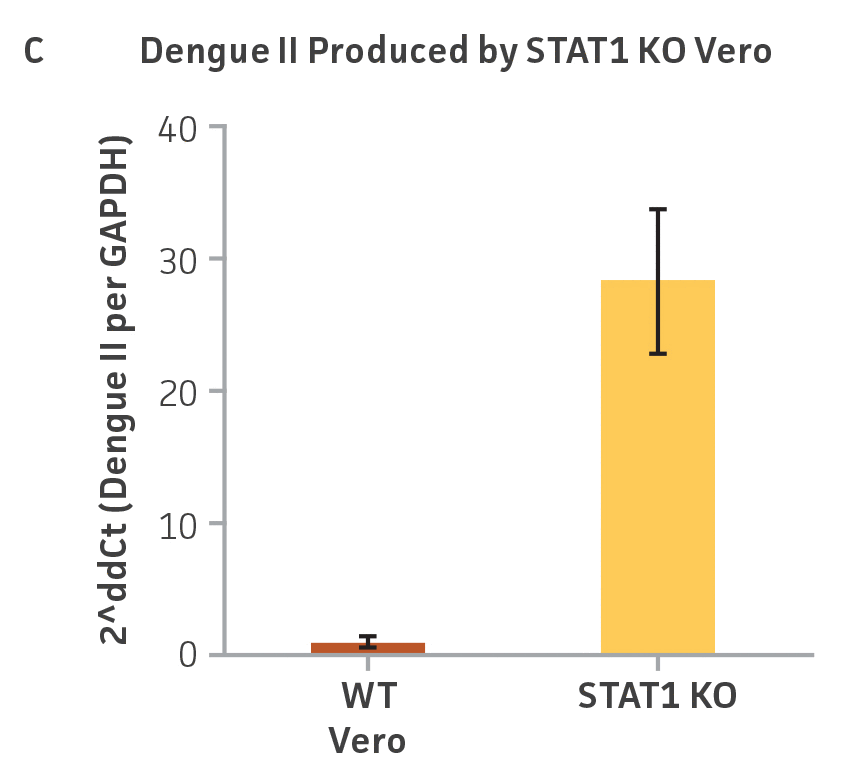
Figure 2: Dengue II virus type 2 strain New Guinea C titer comparison. Vero parental cells and Vero.STAT1 KO cells were seeded and infected with Dengue II New Guinea C virus. Viral supernatants were harvested at day 7 post infection, and then titered by further infecting WT Vero cells in the indicated serial dilutions. (Figure A) Dengue II viruses within the cells were analyzed by immunofluorescence staining and high content microscopy imaging. (Figure B) The 50% tissue culture infective dose (TCID50) were also calculated (n=9). (Figure C) Additionally, RT-PCR shows 30-fold increase in Dengue II viral genomes produced in STAT1 KO Vero cells compared to the parental Vero cells.
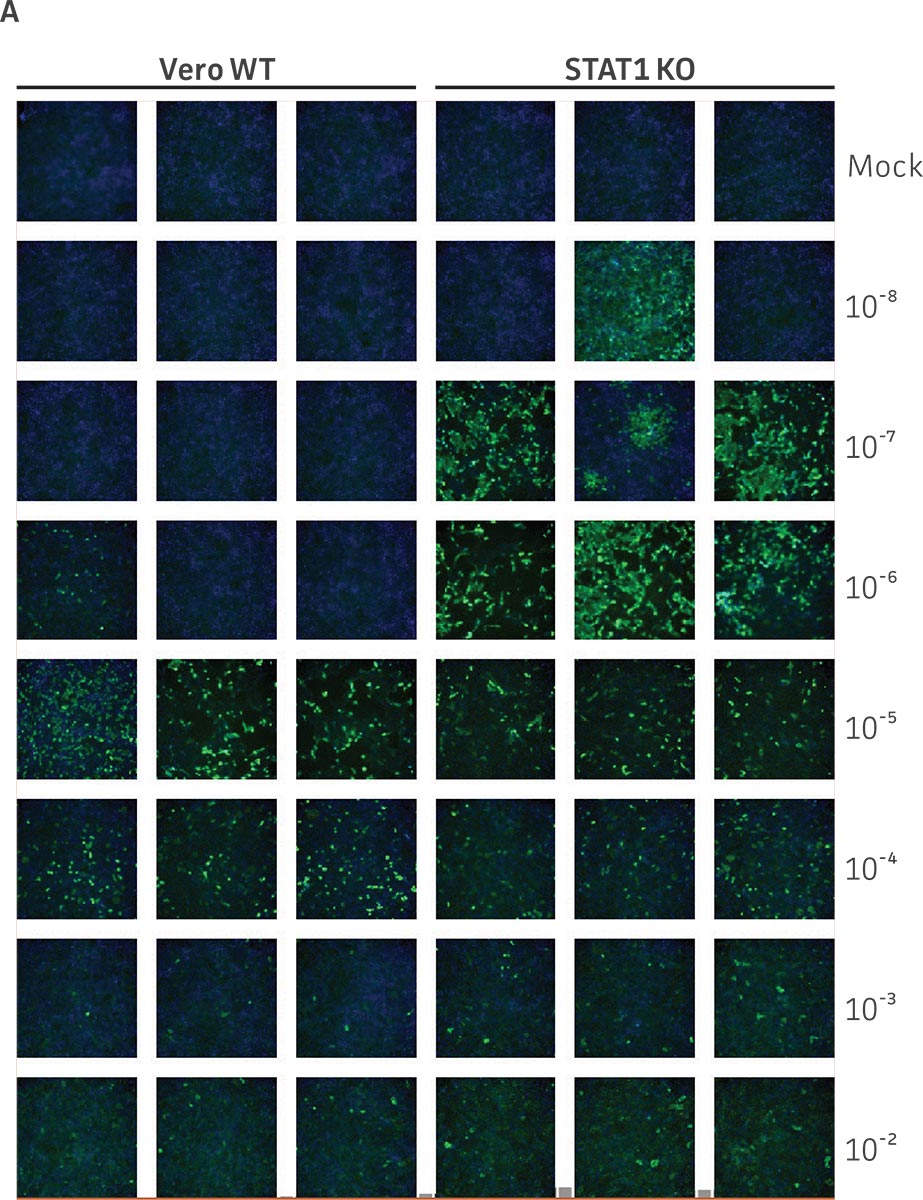
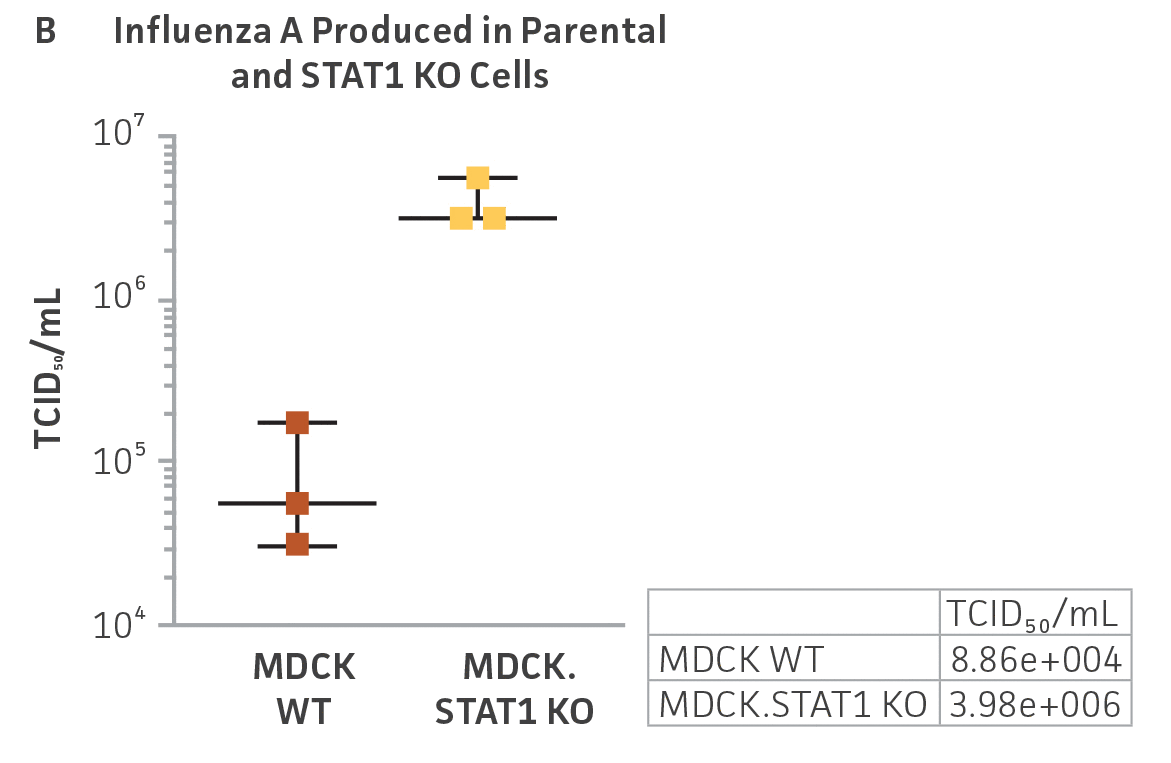
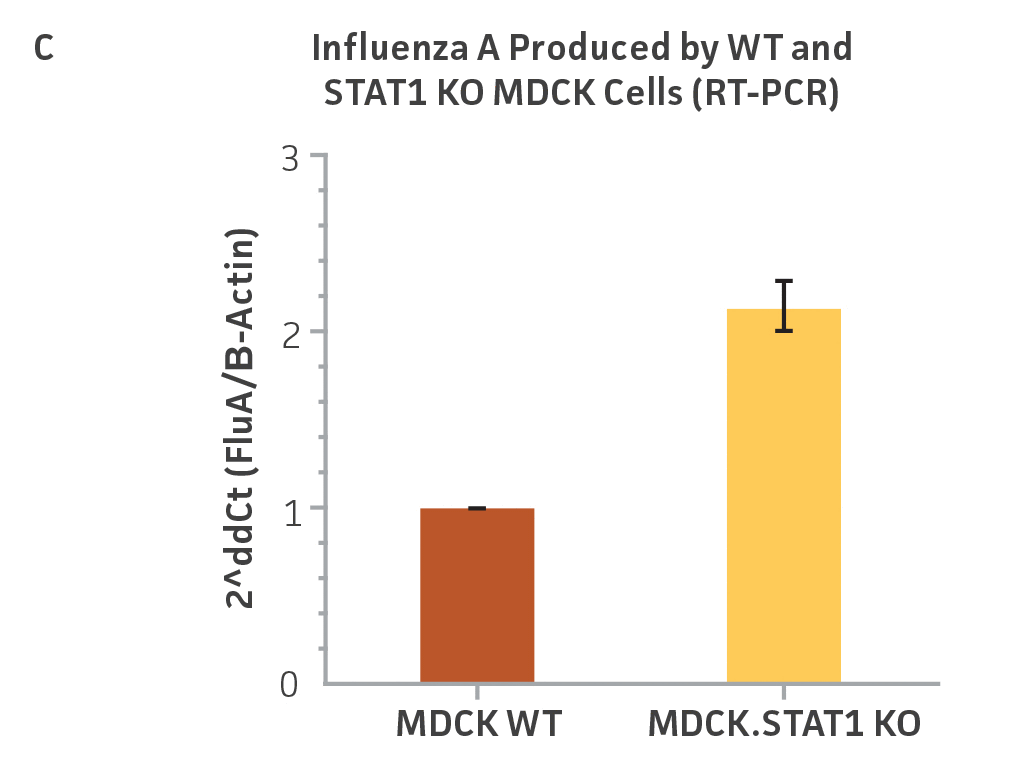
Figure 3: Influenza A titer comparison. Immunostained TCID50 of viral supernatants produced by WT parental and MDCK.STAT1 KO cells. Cells were infected with Influenza A at an MOI of 0.01. Supernatants were collected 48 hours after infection and used to re-infect WT MDCK cells at the indicated dilution. (Figure A) Green – anti-Influenza A stain, Blue – nuclear stain. Cells were infected with Influenza A at an MOI of 0.01 for 48h, then (Figure B) calculated TCID50 of Influenza A viral supernatants produced in MDCK.STAT1 KO cells at 48h post infection (n=3). (Figure C) Additionally, RT-PCR quantification of Influenza A viral shows a 2-fold increase in Influenza A genomes produced in MDCK.STAT1 KO cells compared to the parental cells at 48h post-infection.
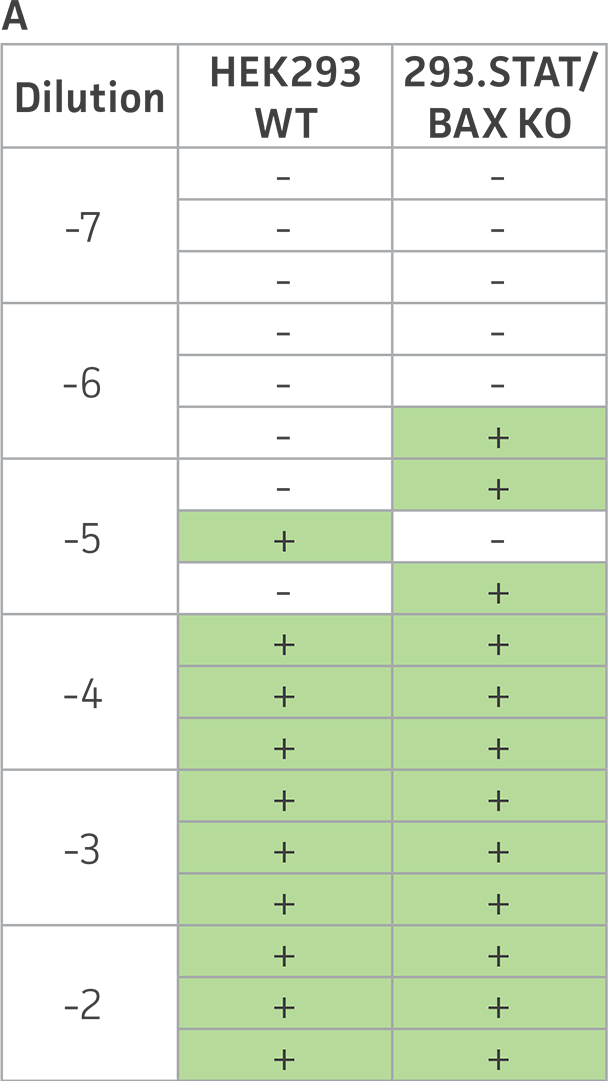
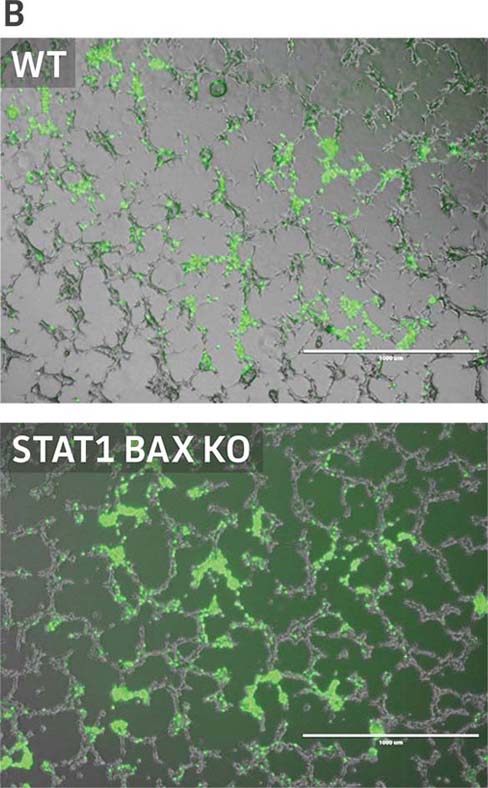
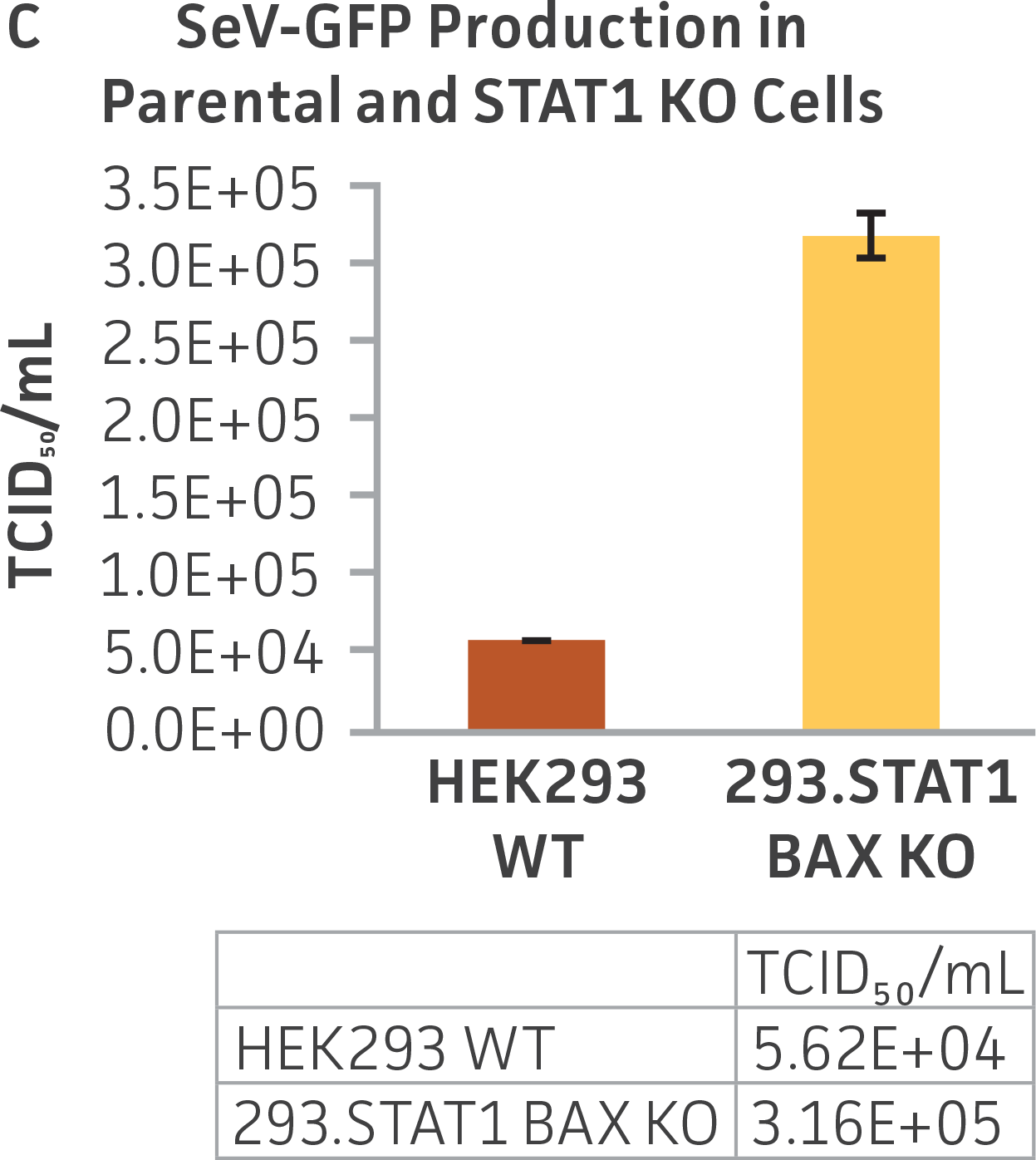
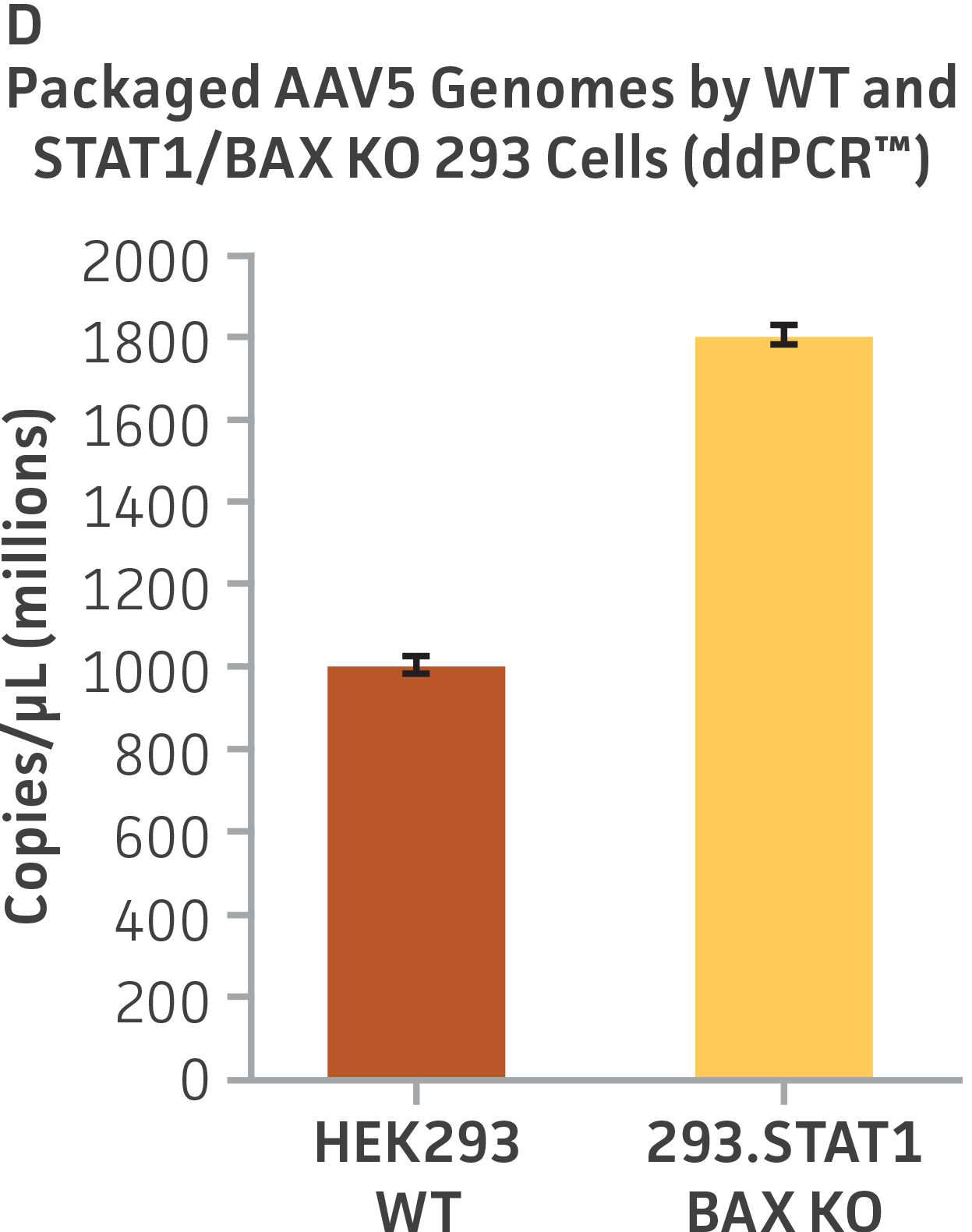
Figure 4: Sendai virus (SeV) and AAV5 titer comparison. (Figure A) TCID50 of GFP SeV supernatants produced by WT parental and 293.STAT1 BAX KO cells. Cells were infected with GFP SeV at an MOI of 0.01. Supernatants were collected 48 hours after infection and used to reinfect WT HEK 293 cells at the indicated dilution. (Figure B) Cells were imaged 48 hours post GFP SeV infection, Scale bar=1000 μm. (Figure C) TCID50 of GFP SeV supernatants produced at 48h post infection were calculated (n=3). (Figure D) Cells were transfected with AAV5 viral vector. The supernatants were collected 48 hours after transfection and used to re-infect WT HEK 293 cells. Droplet digital PCR quantification of AAV5 viral genomes indicated a 1.8-fold increase in viral genomes produced in 293.STAT1 BAX KO cells compared to the parental cells at 72h post-infection.
Download a PDF of this application note
Download NowReferences
- Horvath CM, Darnell JE Jr. The antiviral state induced by alpha interferon and gamma interferon requires transcriptionally active Stat1 protein. J Virol 70: 647-650, 1996. PubMed: 8523587
- Meraz MA, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell 84: 431-442, 1996. PubMed: 8608597
- Cuconati A, et al. Bak and Bax function to limit adenovirus replication through apoptosis induction. J Virol 76(9): 4547-4558, 2002. PubMed: 11932420