Authors: Juan Lopera, PhD;1 Briana Benton, BS;1 Jung-Woo Sohn, PhD;1 Stephen King, MS;1 Tasha M. Santiago-Rodriguez, PhD;2 Emily B. Holister, PhD;2 Matthew C. Wong, BS;3 Nadim Ajami, PhD;3 and Cara Wilder, PhD1
1ATCC, Manassas, VA 20110
2Diversigen, Houston, TX 77021
3Baylor College of Medicine–Alkek Center for Metagenomics and Microbiome Research, Houston, TX, 77030
Introduction
Development of Mock Viral Communities
Using the Whole Virus Standard
Using the Nucleic Acid Virome Standard
Conclusions
References
Abstract
ATCC has developed standardized reference materials for virome research, viral diagnostics, and next-generation sequencing assay optimization. These standards were developed as whole virus and nucleic acid preparations comprising 6 clinically relevant human viral pathogens of diverse genome type, size, organization, and envelope properties. Here, we describe the development and quality control of the ATCC virome standards and present application data demonstrating their use throughout the different stages of a typical virome analysis workflow.
Download a PDF of this application note
Download NowIntroduction
The natural human virome is the collection of eukaryotic viruses, bacteriophages, and endogenous retroviruses found in and within the human body. While it is recognized that the human virome plays an important role in human health and disease, the composition and function of this community, as well as the interactions that occur between viral species and host cells are not well known. By understanding these complex dynamics, researchers are offered a powerful tool for uncovering pathogenesis mechanisms and developing novel preventive and therapeutic applications.1-3
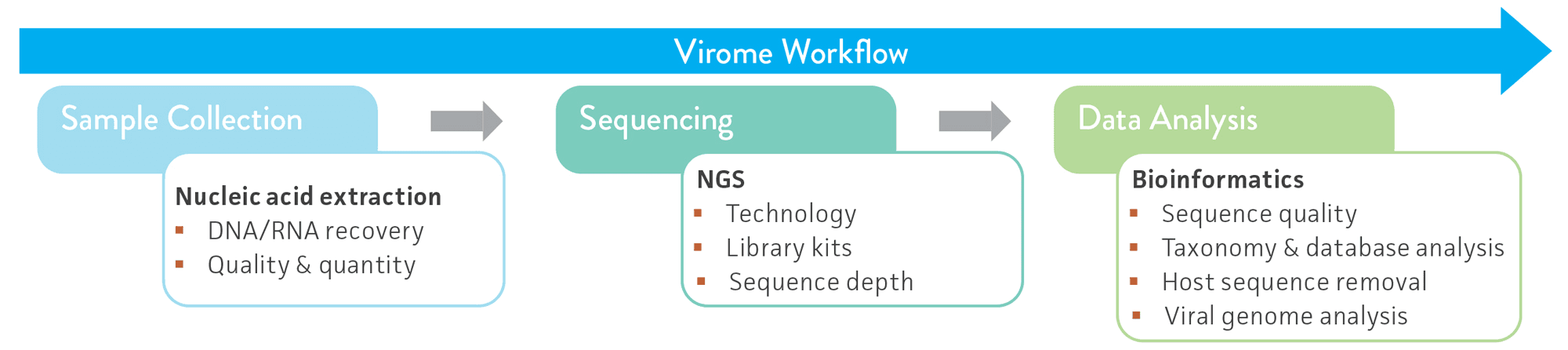
Next-generation sequencing (NGS) technologies have enabled shotgun metagenome sequencing of microbial communities on a large scale at an affordable cost, and they have become the gold-standard method used for virome analyses and molecular diagnostics.4-5 However, despite the significant advancements in these technologies and related data analysis tools, virome research is still challenged by the lack of phylogenetically conserved regions in viruses (akin to the 16S rRNA gene typically used for bacterial and archaeal taxonomic classification), low abundance of viral genome copies, viral sequence variability, and host sequence diversity and abundance.1,6-9 The inherent biases that may arise at each stage of the NGS virome analysis workflow further complicate these issues (Figure 1).
To address these challenges, ATCC created whole virus (ATCC MSA-2008) and nucleic acid (ATCC MSA-1008) virome standards that can be used for assay standardization and reproducibility, or as benchmarks for analysis tools. In the following study, we describe the development and validation of these standards and we present application data from several proof-of-concept studies performed by ATCC and 2 external laboratories. Our data demonstrate that virome standards provide a valuable resource for the scientific community that enable extensive benchmarking and comparative evaluation of different sampling methods, NGS approaches, and bioinformatics tools currently used in virome and diagnostic research.
Development of mock viral communities
ATCC virome standards are fully sequenced, characterized, and authenticated mock viral communities that were developed as whole virus or nucleic acid preparations. These standards comprise 6 viral families (Orthomyxoviridae, Flaviviridae, Pneumovirinae, Reoviridae, Adenoviridae, and Herpesviridae) that were selected on the basis of their clinical relevance and diverse characteristics including genome size, genome type (DNA or RNA), and virion structure (Table 1).
Human and animal cell lines were used to propagate, isolate, and purify whole viruses; nucleic acids (DNA and RNA) were isolated from a preparation of cell lysate and supernatant from specific cell lines Table 1). Following individual viral purification and DNA and RNA isolation and quantitation, whole virus and nucleic acid preparations were mixed in equal proportions based on the genome copy number; the whole virus mix contains approximately 2 × 103 genome copies/μL per virus, and the nucleic acid mix contains approximately 2 × 104 genome copies/μL per virus. Genome copy number was measured via specific fluorescent probes and Droplet Digital PCR (ddPCR; Bio-Rad). After production, both virome standards were evaluated by ddPCR to confirm that all 6 viruses could be detected and that the genome copy number for each virus was close to the expected value (Figure 2). The difference between the observed and expected genome copies for the whole virus standards could be due to differences in viral structure, genome length, or the number of genome segments, which affect nucleic acid extraction and amplification efficiency (Table 1).
Table 1. Selection attributes for strains included in the ATCC Virome Standards.
| Virus name | ATCC number | Genome type | Host (ATCC number)* | Virion structure | Reference GenBank ID | Genome size (Kbp) | Relevance |
|---|---|---|---|---|---|---|---|
| Human herpesvirus 5 | VR-538 | ds DNA | MRC-5 (CCL-171) | Enveloped | X17403.1 | 229.4 | Ubiquitous infection in adult humans, and significant pathogen within immunocompromised populations10 |
| Human adenovirus 40 | VR-931 | ds DNA | HEK-293 (CRL-1573) | Unenveloped | NC_001454.1 | 34.2 | Human gastrointestinal infection and severe infection in children and immunocompromised patients11 |
| Influenza B virus B/Florida/4/2006 | VR-1804 | ss (-) RNA (8 segments) | SPF embryonated chicken eggs | Enveloped | CY018365.1-CY018372.1 | 14.2 | Causes worldwide human epidemics of influenza with high rates of illness and death12 |
| Zika virus | VR-1838 | ss (+) RNA | Vero (CCL-81) | Enveloped | KX830960.1 | 10.8 | Mosquito-borne viral infection that can cause congenital microcephaly in fetuses and infants13 |
| Human respiratory syncytial virus | VR-1540 | ss (-) RNA | HEp-2 (CCL-23) | Enveloped | KT992094.1 | 15.2 | Causes severe respiratory tract infections in humans14 |
| Reovirus 3 | VR-824 | ds RNA (10 segments) | LLC-MK2 Derivative (CCL-7.1) | Capsids | HM159613.1-HM159622.1 | 23.6 | Human respiratory and gastrointestinal infection; oncolytic virus15,16 |
*ATCC cell line used for viral propagation and nucleic acid isolation.
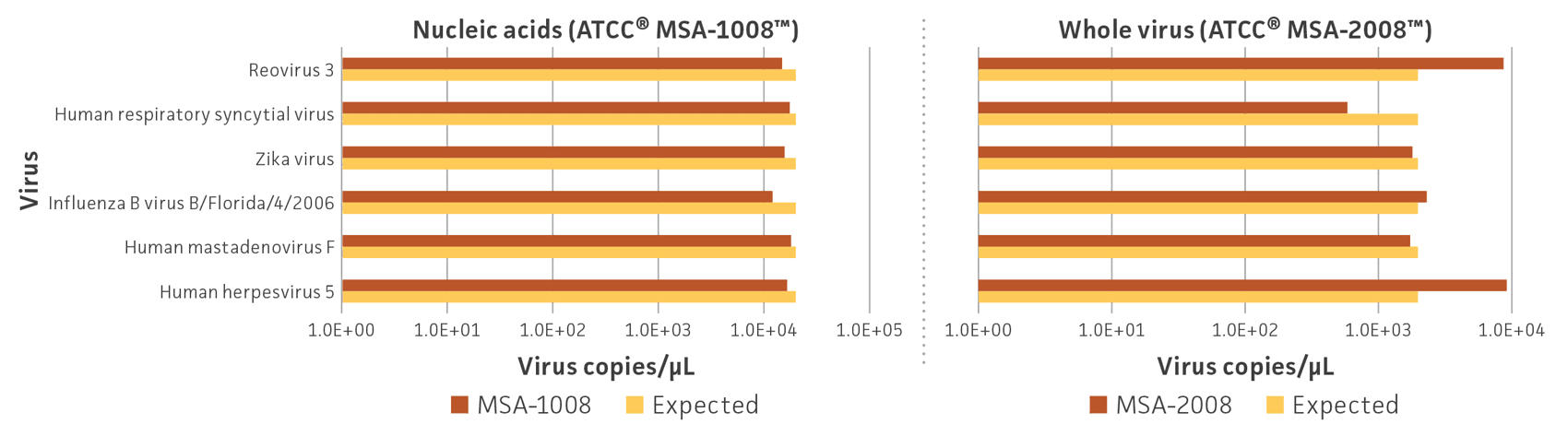
Figure 2. Quantification of estimated genome copy number for each virus in the (A) nucleic acid (ATCC MSA-1008) and (B) whole virus (ATCC MSA-2008) standards. Primers and probes were designed for individual strains and ddPCR assays were performed in triplicate with 3 different dilution factors. Data represent the mean genome copies.
Using the whole virus standard to evaluate and standardize virome workflow methodologies
To demonstrate the application of virome standards in the evaluation and standardization of a full virome analysis workflow, we compared the results obtained from 2 different laboratories that processed and analyzed the whole virus virome standard (ATCC MSA-2008). To remove the biases associated with variations between data analysis platforms, we analyzed and compared the sequence data obtained from both laboratories with the same bioinformatics program: VirMAP (developed at Baylor College of Medicine).7 VirMAP offers an efficient and accurate solution to determine the correct taxonomy and viral abundance from a complex sample by using both nucleotide and protein information to taxonomically classify viral NGS data following individual viral genome reconstruction.7
The results produced from Lab 1 and Lab 2 demonstrated that both analysis workflows were able to detect all 6 viral species in the whole virus mix. However, the viral abundance significantly differed between the 2 studies (Figure 3). Lab 1 used a strategy that included independent steps for library preparation, sequencing, and analysis of DNA and RNA viruses; this strategy produced viral abundances close to the expected values (Figure 3A). In contrast, Lab 2 used the virome standard to design and standardize a different approach that included the use of a single library kit, sequencing run, and analysis to efficiently evaluate both DNA and RNA viruses in the same preparation; this strategy detected less of the RNA viruses as compared to the DNA viruses. The variability observed between the 2 laboratories could be due to the use of different nucleic acid extraction kits, library kits, or sequencing methodology. These results demonstrate the need and utility of the whole virus virome standard (ATCC MSA-2008) as a reference material for facilitating the design and optimization of each stage of a virome workflow.
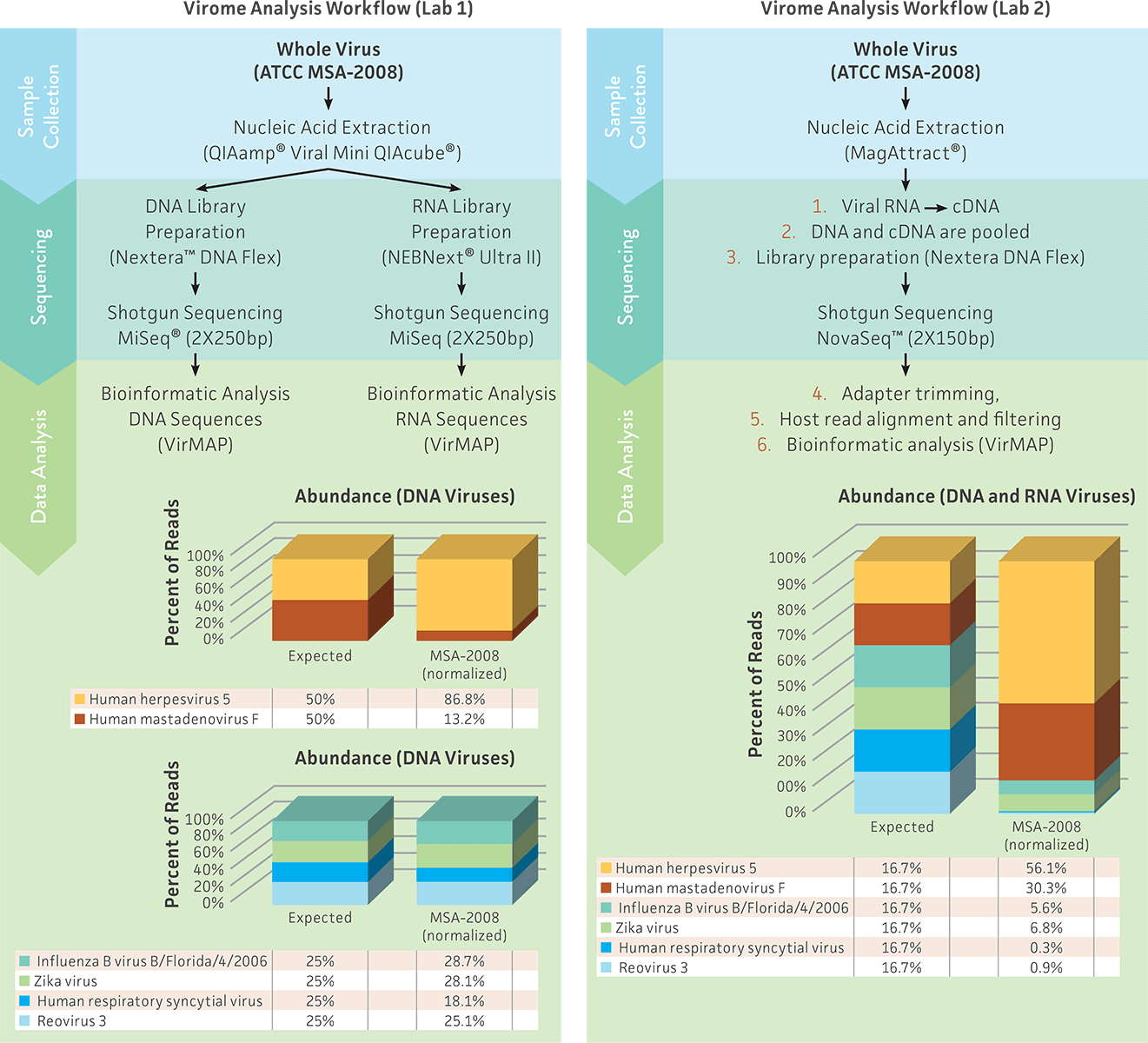
Figure 3. Comparison of 2 different experimental approaches for evaluating nucleic acid extraction, library preparation, sequencing, and data analysis by using the whole virus virome standard (ATCC MSA-2008). The sequence files obtained from the 2 laboratories were analyzed using VirMAP.7 The total viral reads that were classified as viruses were normalized by individual viral genome size (Table 1); the individual viral abundance is presented as the normalized percent of read.
Using the nucleic acid virome standard to evaluate data analysis tools for viral metagenomic studies
A critical step and major challenge in virome data analysis is the selection of the right bioinformatic tools and viral reference databases required to effectively identify viral sequences within a complex mixture of other microorganism- and host-associated sequences.7,8 The removal of contaminating reads originating from the host and other organisms through bioinformatics is a frequently used approach that typically focuses on mapping and filtering reads based on their alignment against reference sequences. However, this approach is limited by the availability of known and precise reference genomes.17,18 To that end, we used ATCC virome standards to benchmark and compare virome analyses by using 2 bioinformatics tools (VirMAP and Autometa) that incorporate innovative approaches that do not require known reference sequences. Briefly, using the nucleic acid virome standard (ATCC MSA-1008) and a similar approach evaluated for Lab 1 (Figure 3), we created separate DNA and RNA libraries and produced independent DNA and RNA shotgun metagenome sequences. We then analyzed the same datasets with the 2 bioinformatic tools. As described above, the VirMAP pipeline uses an approach based on information from nucleotide and protein databases for taxonomic classification of NGS reads and individual viral genome reconstruction.7 In contrast, Autometa is a metagenome binning pipeline that splits de novo–assembled contigs into kingdom bins, allowing for the separation of virus contigs from other microorganism and host-derived sequences by examining the lowest common ancestor (LCA) of predicted proteins in each contig to assign the taxonomic classification.19
Bioinformatic analysis using VirMAP efficiently separated host-associated reads from viral sequences and enabled the calculation of individual viral abundance for both DNA and RNA datasets obtained from the nucleic acid virome standard (ATCC MSA-1008). For shotgun DNA sequencing, 34.2% (9.0 million) of the total reads (26.4 million) were specific to the 2 DNA viral genomes, and the remaining 65.8% (17.4 million) of reads were associated with DNA from the host and other microorganisms. For RNA sequencing, 8.4% (3.3 million) of the total reads (39.2 million) were specific to the 4 RNA viruses, and the remaining 91.6% (35.9 million) were associated with RNA from the host and other microorganisms (Figure 4).
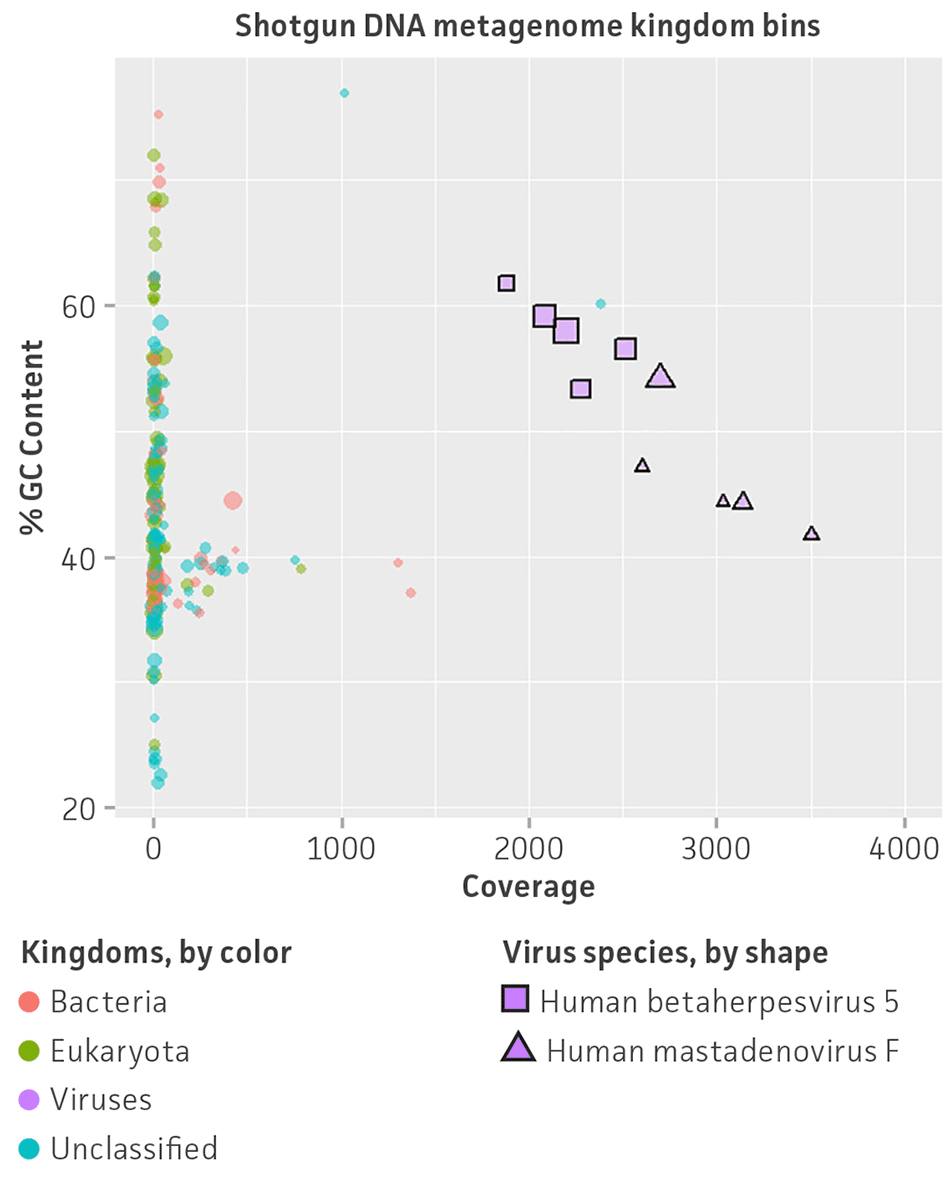
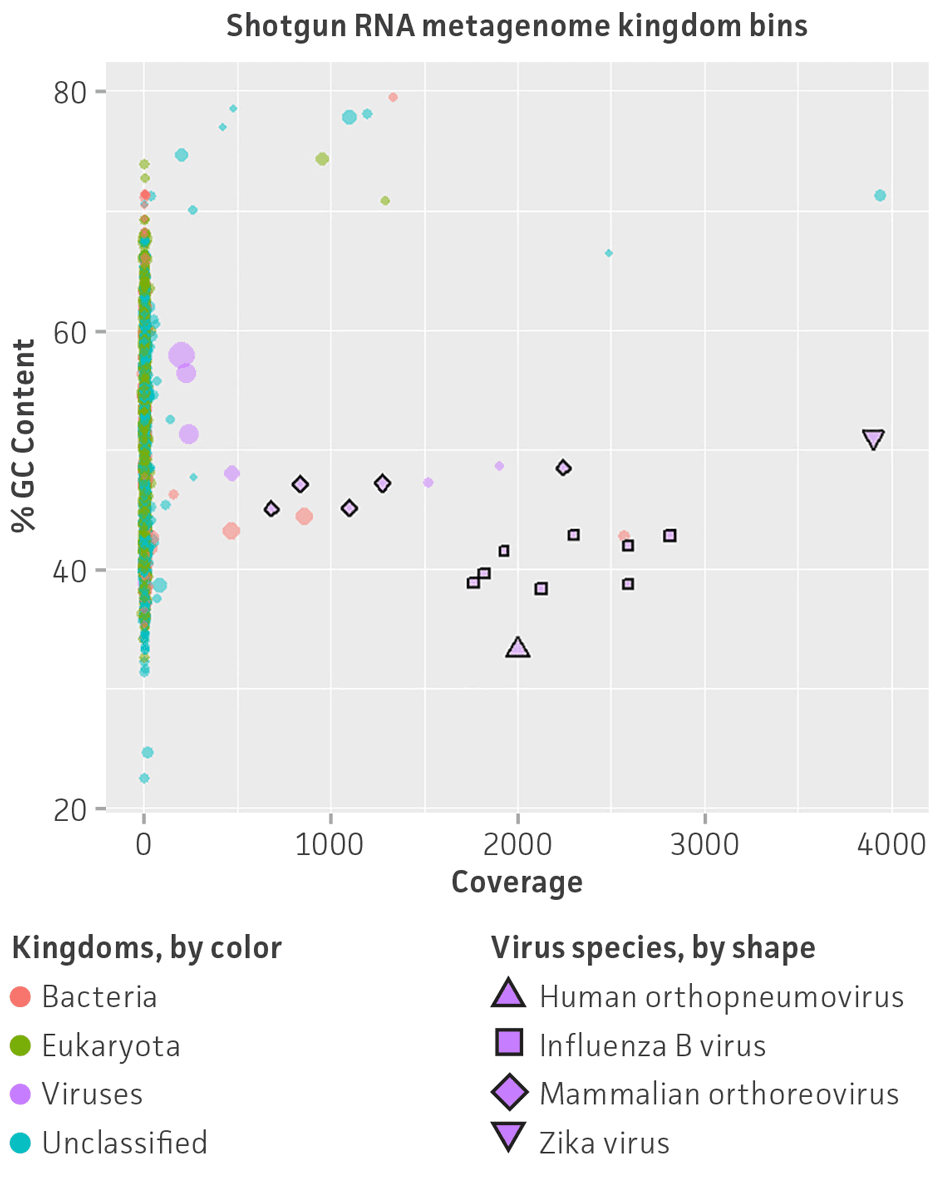
Regarding viral abundance, results from VirMAP showed individual values close to the expected percentages. Here, shotgun DNA sequencing showed a relative abundance of 46.3% for herpesvirus and 53.7% for adenovirus, and shotgun RNA sequencing showed a relative abundance of 38.3% for Zika virus, 19.7% for human respiratory syncytial virus, 20.8% for reovirus, and 21.8% for influenza B virus (Figure 4). Similar to VirMAP, bioinformatic analysis using Autometa was able to properly taxonomically classify DNA and RNA viral sequences from the nucleic acid virome standard (Figure 5). Additionally, both Automata and VirMAP were able to separate individual viral genomes, which enabled the evaluation of NGS depth, genome assembly completeness, and other important viral metagenome measures that researchers need in order to facilitate the standardization and optimization of diagnostic and functional characterization of complex virome samples (Figure 5). Together, these results exemplify and demonstrate the utility of the nucleic acid virome standard in benchmarking, comparing, and selecting an appropriate bioinformatics tool for viral microbiome analysis while also enabling the optimization of NGS parameter runs for the complete genome characterization of virome samples.
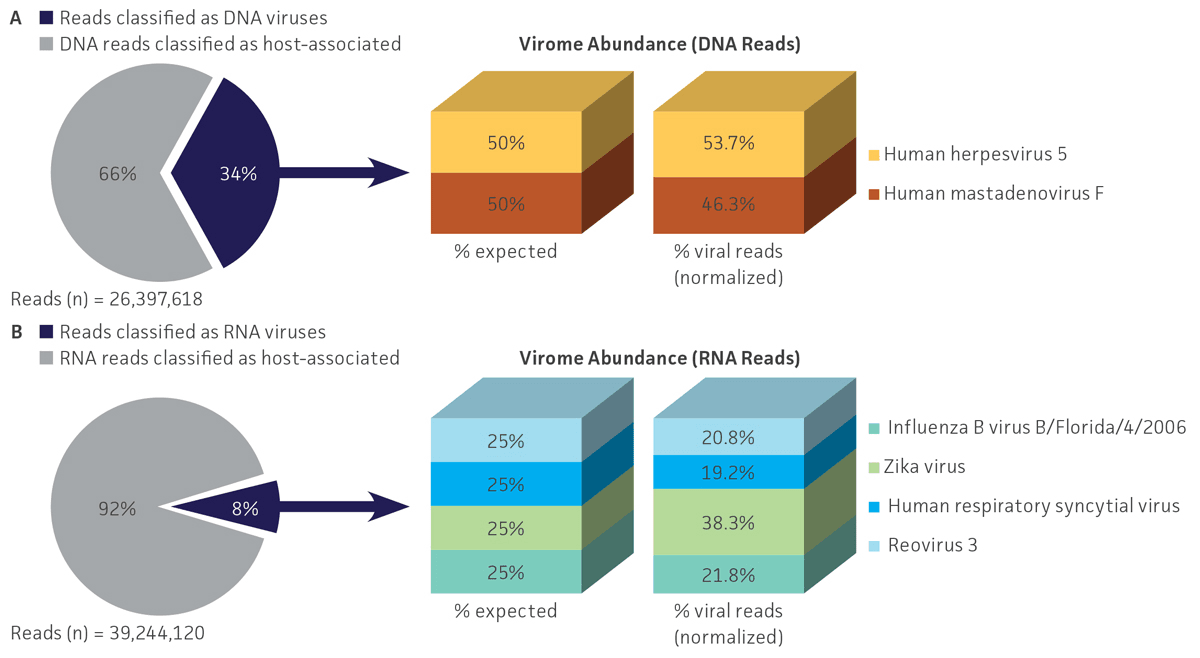
Figure 4. Shotgun metagenome profiling of the nucleic acid virome standard (ATCC MSA-1008). VirMAP analysis results from (A) Illumina DNA shotgun sequencing and (B) Illumina RNA shotgun sequencing of the standard showed efficient host-associated and viral sequencing separation, and the individual viral profiling in the mix was close to the expected abundances.
|
|
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
1 = Expected based on the reference GenBank ID from Table 1.
2 = contigs ≥1 kbp
3 = Estimated completeness [(number of CDS estimated in reference GenBank / number of CDS estimated in individual viral bin) × 100]. Number of CDS were estimated using Prokka software tool
Figure 5. Shotgun metagenome binning analysis of the nucleic acid virome standard (ATCC MSA-1008). Autometa analysis results and individual viral genome assembly measured from (A) Illumina DNA shotgun sequencing and (B) Illumina RNA shotgun sequencing of the virome standard. Metagenomic binning plots represents 2-dimensional clustering (%GC and coverage components) of individual de novo assembly contigs (colored spots represent kingdom bins and stars show viral bins).
Conclusions
Overall, these proof-of-concept studies highlight the utility and importance of virome standards in the optimization of sample processing, sequencing, and bioinformatics methods throughout a virome analysis workflow. Due to the complexity of virome sample processing and sequencing, significant challenges can be posed by biases introduced during sample preparation, nucleic acid extraction, library preparation, sequencing, or data interpretation. By incorporating standards in the assay development and validation processes, researchers are offered a comprehensive solution for standardizing data from a wide range of sources and generating consensus among viral microbiome studies.
Acknowledgments
We would like to thank Dr. Jason Kwan at the University of Wisconsin-Madison for his support during the installation and application of the Autometa pipeline.
Download a PDF of this application note
Download NowReferences
- Santiago-Rodriguez TM, Hollister EB. Human Virome and Disease: High-Throughput Sequencing for Virus Discovery, Identification of Phage-Bacteria Dysbiosis and Development of Therapeutic Approaches with Emphasis on the Human Gut. Viruses 11(7): pii: E656, 2019.
- Beller L, Matthijnssens J. What is (not) known about the dynamics of the human gut virome in health and disease. Curr Opin Virol 37: 52-57, 2019.
- Carding SR, Davis N, Hoyles L. Review article: the human intestinal virome in health and disease. Aliment Pharmacol Ther 46(9): 800-815, 2017.
- Gu W, Miller S, Chiu CY. Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection. Annu Rev Pathol 14: 319-338, 2019.
- Datta S, et al. Next-generation sequencing in clinical virology: Discovery of new viruses. World J Virol 4(3): 265-276, 2015.
- Lambert C, et al. Considerations for Optimization of High-Throughput Sequencing Bioinformatics Pipelines for Virus Detection. Viruses 10(10): pii: E528, 2018.
- Ajami NJ, et al. Maximal viral information recovery from sequence data using VirMAP. Nat Commun 9(1): 3205, 2018.
- Sutton TDS, et al. Choice of assembly software has a critical impact on virome characterisation. Microbiome 7(1): 12, 2019.
- Wylie KM, Weinstock GM, Storch GA. Emerging view of the human virome. Transl Res 160(4): 283-290, 2012.
- Jackson JW, Sparer T. There Is Always Another Way! Cytomegalovirus’ Multifaceted Dissemination Schemes. Viruses 10(7): 383, 2018.
- Chen S, Tian X. Vaccine development for human mastadenovirus. J Thorac Dis 10(Suppl 19): S2280-S2294, 2018.
- Krammer F, et al. Influenza. Nat Rev Dis Primers 4(1): 3, 2018.
- Musso D, Ko AI, Baud D. Zika Virus Infection–After the Pandemic. N Engl J Med 381(15): 1444-1457, 2019.
- Rossey I, Saelens X. Vaccines against human respiratory syncytial virus in clinical trials, where are we now? Expert Rev Vaccines 18(10): 1053-1067, 2019.
- Besozzi M, et al. Host range of mammalian orthoreovirus type 3 widening to alpine chamois. Vet Microbiol 230: 72-77, 2019.
- Kemp V, et al. Characterization of a replicating expanded tropism oncolytic reovirus carrying the adenovirus E4orf4 gene. Gene Ther 25(5): 331-344, 2018.
- Miller JR, et al. A host subtraction database for virus discovery in human cell line sequencing data. F1000Res 7: 98, 2018.
- Daly GM, et al. Host Subtraction, Filtering and Assembly Validations for Novel Viral Discovery Using Next Generation Sequencing Data. PLoS One 10(6): e0129059, 2015.
- Miller IJ, et al. Autometa: automated extraction of microbial genomes from individual shotgun metagenomes. Nucleic Acids Res 47(10): e57, 2019.