Authors: Juan Lopera, PhD; Andrew Frank, MS; Anna McCluskey BS; Stephen King, MS; Samantha Fenn, BS; Karin Kindig, MS; Marco Riojas, PhD; Jung-Woo Sohn, PhD; Holly Sadural, BS; Benton Briana, BS; and Cara Wilder, PhD
Introduction
Survey of ATCC Genome Sequences in Public Genome Databases
Evaluation of ATCC Genome Sequences From Public Databases
Development and Evaluation of Hybrid De Novo Bacterial Assembly
Summary
References
Acknowledgments
Abstract
While recent technological advancements have enabled the generation of vast amounts of whole-genome sequencing data, publicly available reference genomes often lack quality, completeness, authenticity, accuracy, and traceability. The reliability of these data is further called into question as they may have been generated using untraceable cultures and older methodologies. In the following study, we surveyed the status of ATCC bacterial genome sequences in public databases and described the implementation of a genome sequencing workflow designed to provide reference-quality whole-genome sequences that are derived from authenticated ATCC materials.
Download a PDF of this application note
Download NowIntroduction
The advancement and accessibility of next-generation sequencing (NGS), cloud computing, and sequence analysis tools have rapidly transformed microbiological research by opening up applications in the areas of clinical diagnostics, drug discovery, public health, microbiome research, antimicrobial resistance studies, and industrial and environmental microbiology.1-3 Many of these NGS-based applications have relied on the availability of high-quality assembled and annotated genome sequences in public databases to serve as references and control for bioinformatic analyses.4-6 However, despite the large number of existing microbial genome sequences in various public databases, the quality, completeness, authenticity, accuracy, and traceability of some genomic data are frequently questionable as they could have been generated by various researchers using non-authenticated cultures and older sequencing and analysis technologies. Further, the lack of standardized methodologies for best practices during the sequencing and assembly of reference genomes exacerbates the underlying problems.
For researchers to accurately interpret their results and make insightful correlations with in silico models, it is essential that they have access to reliable genomic information tied back to authenticated, fully characterized materials of known and reliable provenance. Therefore, as part of our initiative to enhance the authentication of our products, we have identified the key challenges regarding existing microbial genome databases and have developed a solution for improving the quality of reference genome sequences. In the following study, our results demonstrated that there is no significant representation of ATCC strains with genome sequences available in public databases, and even fewer of these strains have complete circularized chromosome and plasmids. Here, we examine those results in detail and discuss the development and application of our standardized end-to-end sequencing and assembly workflow for producing reference-quality genome sequences (Figure 1).
![]()
Figure 1. Comprehensive ATCC bacterial whole-genome sequencing workflow.
Survey of ATCC genome sequences in public genome databases
A reference genome is a high-quality sequence published in a database that provides a representative example of a species; these sequences are reviewed and validated extensively.7,8 Today, there are multiple distinct microbial genomic resources, tools, and databases publicly available in internet portals.9-13 In this study, we focused the survey of bacterial genome sequences on those identified by the depositor of the sequence as ATCC materials in two important public genome sequence databases: Microbial Genomes (NCBI-NIH) and Ensembl Bacteria (EMBL-EBI). The purpose of the study was to collect information about the number and assembly status (scaffold, contigs, and complete bacterial chromosomes and plasmids) of published ATCC strains in two of the most frequently used databases that are of interest for microbiology research (Table 1). Here, database records containing the keyword “ATCC” were extracted and compared to the entire collection of database entries. Records in the Microbial Genomes database provided the complete information of assembly level (contigs, scaffolds, chromosomes, and plasmids) (Table 1). Since records in the Ensembl Bacteria database do not directly present the information with assembly level information, a Python script that extracts the genome assembly statistics based on the accession number was written and run to generate the data (Table 1).
Of the records evaluated in the public databases, we identified a total of 1,807 (1.1%) ATCC prokaryote genomes sequences classified as RefSeq in the Microbial Genomes database and 715 (1.6%) ATCC strains in the Ensembl Bacteria database (Table 1). Further, we identified that the surveyed genome sequences were primarily characterized to be incomplete genome drafts consistent of multiple noncontiguous scaffolds or contigs. Specifically, 27.7% of ATCC strains in the Microbial Genomes database and 10.9% ATCC strains from the Ensembl Bacteria database were identified as complete genomes, whereas 72.3% and 89.1% of the ATCC strains in the Microbial Genomes and Ensembl Bacteria databases, respectively, were genome drafts with fragmented genome scaffolds or contigs (Table 1, Figure 2). We also observed that in the Microbial Genomes database approximately 12% of ATCC strains had more than one genome report available. These results demonstrate that while there are a relatively large number of ATCC genome sequences available in multiple public databases, there is a deficiency of complete ATCC circularized genome and plasmids sequences (Table 1, Figure 2).
Table 1. Summary of the microbial genome database survey results
| Database | # of genome sequences (%) | Contigs or scaffolds (%) | Complete genome or chromosome (%) | Genome with plasmids (%) |
|---|---|---|---|---|
| Microbial Genomes (NCBI-NIH) (RefSeq prokaryote database) | 165,807* (80.2%) | 149,171 (90.0%) | 16,636 (10.0%) | 6,333 (3.8%) |
| ATCC strains in Microbial Genomes (NCBI-NIH) (RefSeq prokaryote databases) | 1,807 (1.1%) | 1307 (72.3%) | 500 (27.7%) | 193 (10.7%) |
| Ensembl Bacteria (EMBL-EBI) | 44,011* (96.9%) | 39,203 (89.1%) | 4,808 (10.9%) | NA** |
| ATCC strains in Ensembl Bacteria (EMBL-EBI) | 715 (1.6%) | 521 (72.9%) | 194 (27.1%) | NA** |
*Microbial Genomes database = total 206,660 records evaluated with 165,807 RefSeq hits; Ensembl Bacteria database = 45,438 records evaluated, with 44,048 specific bacteria hits (1,390 record from Ensembl Bacteria database were identified as fungal or viral organisms).
**NA, not applicable; data was not available in the databases
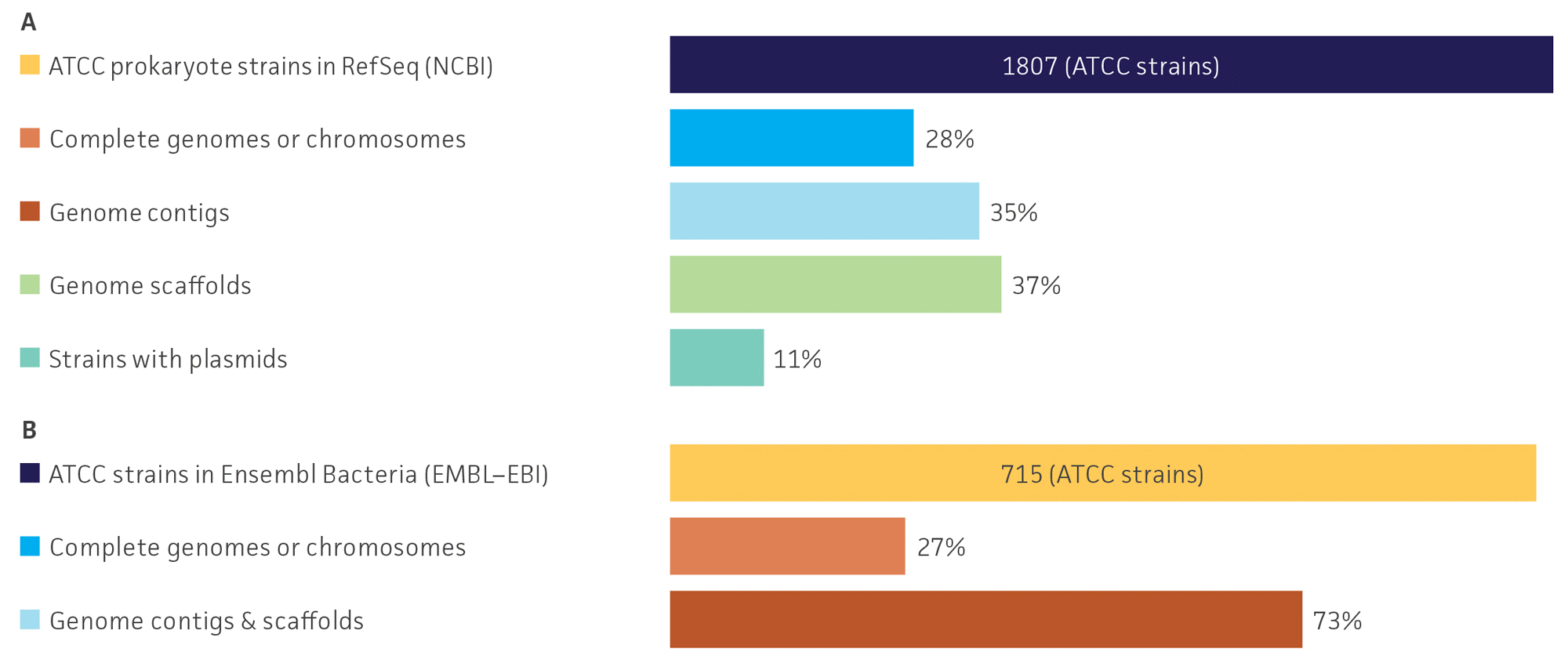
Figure 2. Survey profiles of ATCC genomes organized by assembly status in the (A) Microbial Genomes and (B) Ensembl Bacteria. Note: EMBL-EBI does not differentiate between contigs and scaffolds and does not include plasmid information.
Evaluation of ATCC genome sequences from public databases
To evaluate the quality of published ATCC genomes from public databases and to demonstrate the need of credible reference-quality genomes, we analyzed a select group of strains via sequencing. Here, 100 bacterial strains identified in our genome database survey as having complete assemblies were randomly selected for analysis. Then, using nucleic acids extracted from low passage ATCC bacterial cultures, we re-sequenced the selected strains and analyzed each sequence using customized reference-based assembly (short-read alignment/mapping to published genome sequences) and hybrid de novo assembly (short- and long-read analysis) workflows.
Preparation and quality control of DNA templates
Unlike many of the bacterial genome sequences deposited in public databases, we began our genome sequencing efforts with the comprehensive traceability of ATCC authenticated strains. This allows us to validate the source of the bacterial culture and genomic DNA while linking to vital metadata, thus enabling downstream references and support for analyses. Briefly, before we engaged in the quality assessment of ATCC genomes present in public databases, we carefully reviewed the classification of the bacterial cultures and evaluated the quality and purity of the DNA template used for NGS sequencing. To facilitate the successful NGS library preparation for multiple sequencing platforms (long- and short-read sequences), we used either input DNA obtained directly from authenticated and fully characterized ATCC nucleic acids from our repository or DNA with high molecular weight (NGS-ready DNA) and fragment sizes bigger than 20 kb that were extracted directly from our cultures. The quality and quantity of the DNA used in this study were measured via a DNA analyzer (Agilent) and a fluorescent dye-based method PicoGreen, respectively (Figure 3, Table 2).
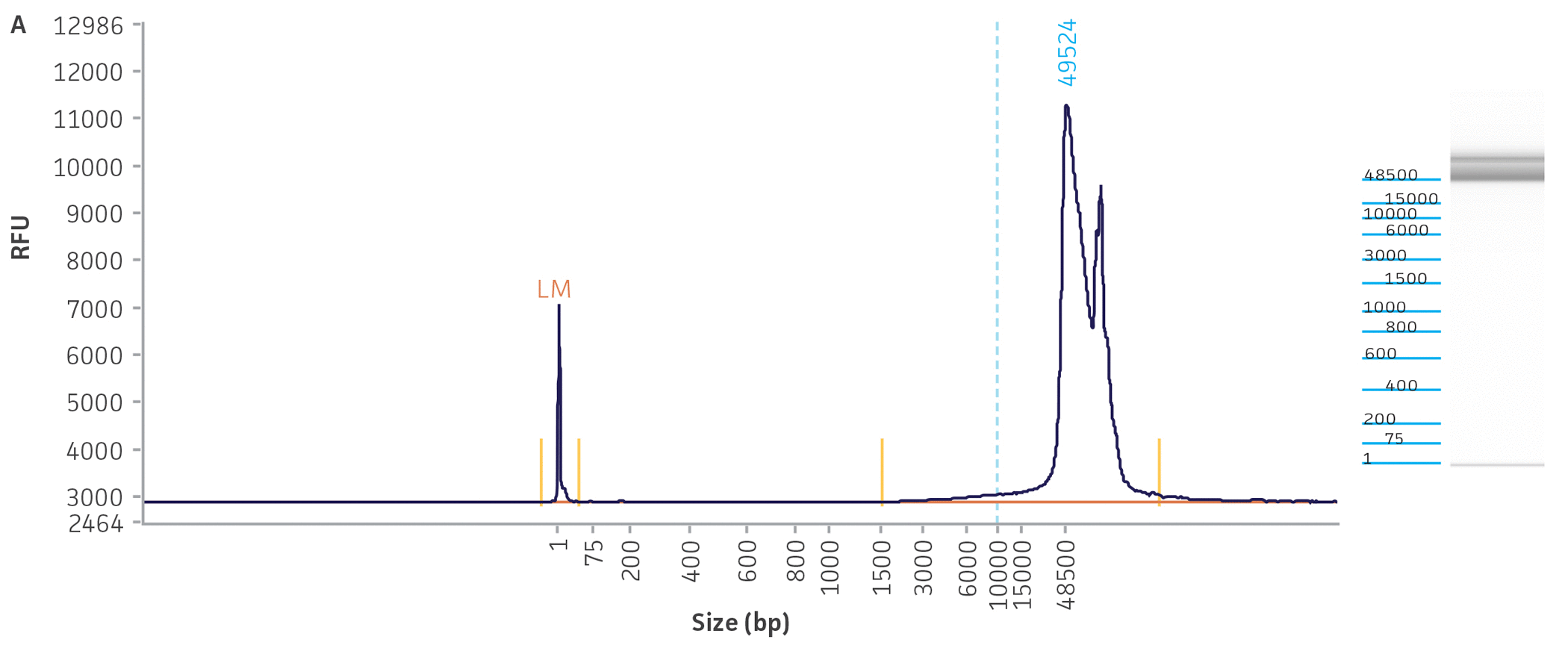
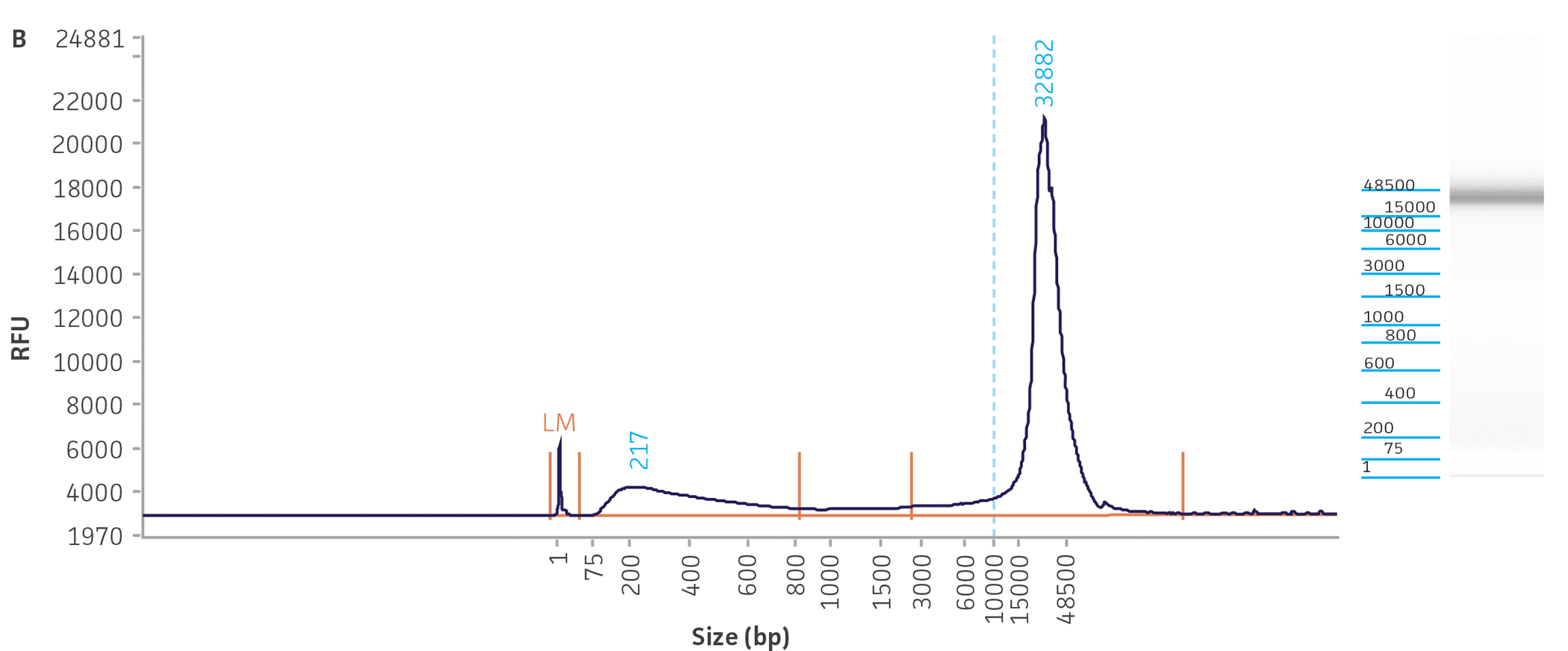
Figure 3. Quality assessment for NGS-ready DNA used in this study. The fragment size graph obtained from the Agilent Fragment Analyzer platform demonstrates the size distribution of total DNA. The graphs depict examples of DNA quality assessments for a gram-negative (A) Escherichia coli (ATCC 8739DX) and gram-positive strain (B) Staphylococcus aureus (ATCC 6538DX), respectively.
Table 2. Summary of DNA quality and quantity measurements before NGS
| ATCC No. | Species | PicoGreen (ng/µL) | A260/A280 | DNA fragment size (range)** |
|---|---|---|---|---|
| 8739DX* | Escherichia coli | 101.9 | 1.92 | 49.5 kb (1.5 – >60 kb) |
| 13048DX* | Klebsiella aerogenes | 98.1 | 1.86 | 49.5 kb (1.6 – >60 kb) |
| 11828DX* | Cutibacterium acnes | 197.7 | 1.84 | 29.8 kb (0.8 – >60 kb) |
| 6538DX* | Staphylococcus aureus | 97.8 | 1.85 | 32.9 kb (2.7 – >60 kb) |
| BAA-2797DX* | Pseudomonas aeruginosa | 153.3 | 1.99 | 44.1 kb (1.1 – >60 kb) |
| 824D-5 | Clostridium acetobutylicum | 73.8 | 2.05 | 12.5 kb (4.6 – 57.8 kb) |
| 6538D-5 | Staphylococcus aureus | 37.1 | 2.00 | 26.2 kb (6.9 – >60 kb) |
| 27774D-5 | Desulfovibrio desulfuricans | 69.2 | 1.99 | 58.5 kb (13.3 – >60 kb) |
| 11842D-5 | Lactobacillus delbrueckii | 64.8 | 2.02 | 41.9 kb (6.1 – >60 kb) |
| 15697D-5 | Bifidobacterium longum | 76.2 | 1.95 | 51.3 kb (10.5 – >60 kb) |
*NGS-ready DNA
**DNA fragment size represents the main peak reported by the fragment analyzer
High-quality NGS sequences
Because NGS has emerged as a sensitive and precise tool for microbial characterization, diagnostics, and discovery, assessing the quality of the raw NGS data has become indispensable for ensuring the credibility of assemblies and the annotation of reference genomes.6,14,15 In public databases, the general submission process for raw sequence data requires some data quality information. Sequence Read Archive (SRA) requires supporting per-base quality scores for all submitted sequences. For the genome assemblies, whole-genome sequencing (WGS) submission requests the base-level quality for which files are not strictly required. However, there are not any standardized sequence quality thresholds that measure or regulate the excellence of the genomic information deposited in public databases.15-17 For this reason, we have developed and implemented a rigorous quality control protocol that includes the analysis of raw sequence quality scores and removal (trimming) of low-quality segments and undefined nucleotides as well as a read-based contamination quality control via the One Codex database (Figure 4). For additional details on the quality control processes we have implemented, see the ATCC Genome Portal Technical Document.
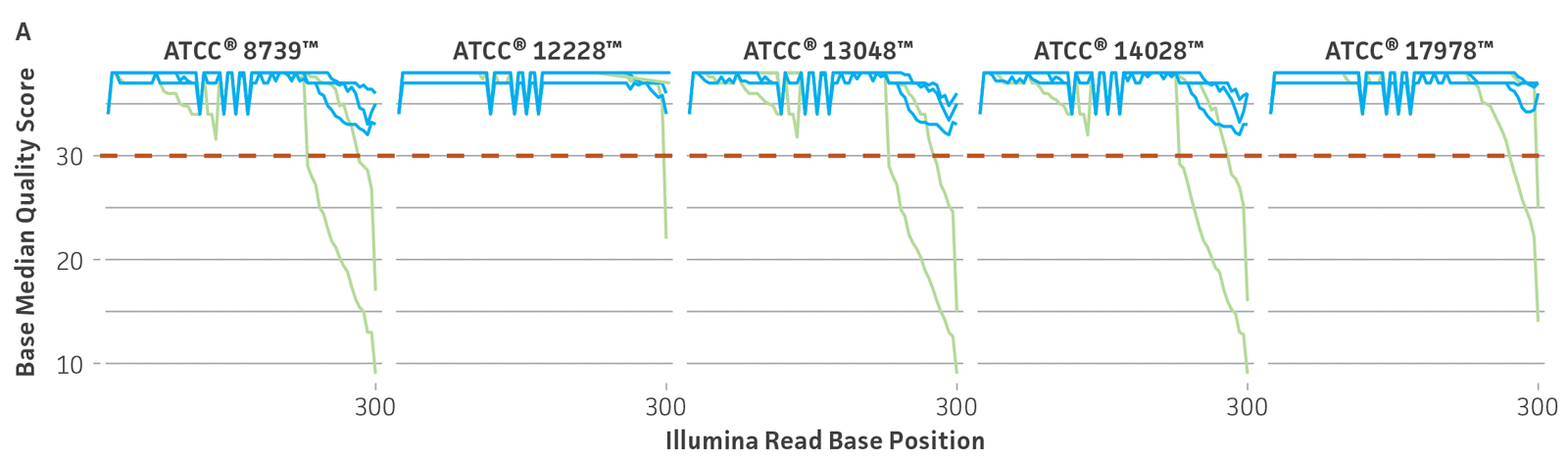
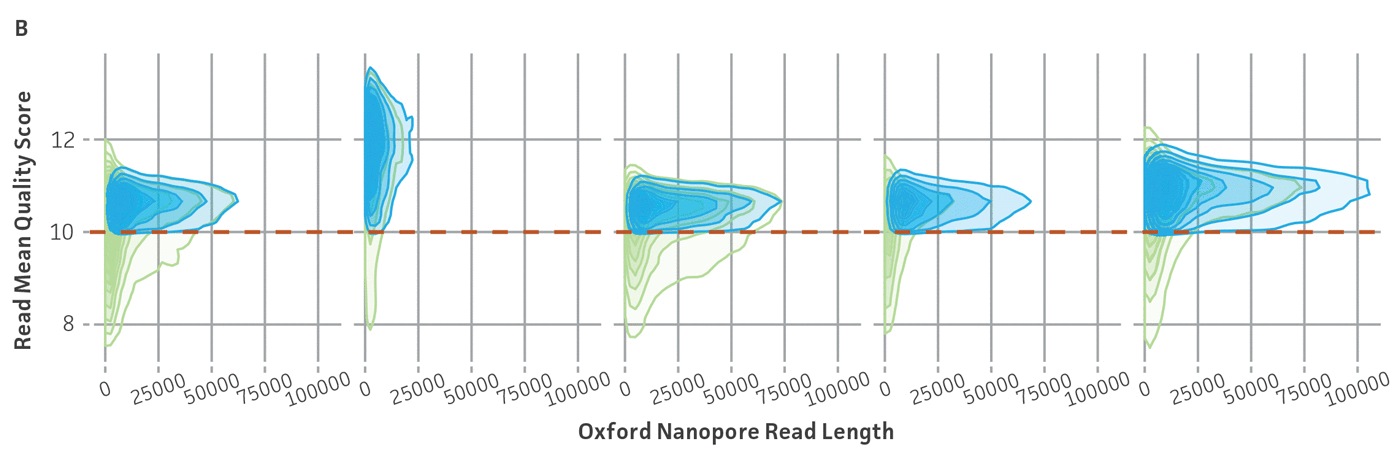

Figure 4. ATCC’s bacterial genome sequencing quality control. The dashed line indicates the quality score cutoff used for each sequencing technology. (A) Quality of Illumina reads. (B) Length distribution of reads from the Oxford Nanopore Technologies (ONT) platform. This approach ensures the longest, highest-quality reads are used for assembly. Thus, the lengths of ONT raw sequence and quality scores were evaluated by measuring read lengths N50 (>5000kb), quality scores (>10), and total yield of sequence runs. (C) Sample composition describes NGS composition by aligning each individual read to a reference database. We use the One Codex microbial genomics platform to perform read-level, k-mer–based taxonomic classification and estimation of strain abundances on our processed Illumina read sets.
Reference-based analysis and variant calling
We evaluated the level of genetic variation between published sequences and NGS sequences obtained directly from ATCC cultures. For 100 sequences identified as ATCC materials in public databases, we ran a reference-based analysis tool on our short reads to identify single nucleotide variations (SNVs) and indels (small insertion/deletion). Briefly, high-accuracy and high-coverage (>100x) Illumina sequences (MiSeq PE 2x300) from ATCC DNAs corresponding to the selected strains were aligned and mapped to published reference assemblies. The genome variants threshold was fixed to a variant average coverage greater than 100x. To validate our results, all of the sequences were first validated by the previously described quality control filter, and then six random strains were sequenced and analyzed in duplicate (Table 4).
Our results demonstrated that approximately 33% of the 100 strains evaluated have fewer than 50 variants (SNVs and indels); 14 strains showed low sequence variation with fewer than 5 variants, and 8 strains showed large sequence variation with more than 500 variants detected. When SNVs and indels were evaluated separately, we found that 18% of the strains exhibited more than 50 SNVs and 37% of the public genomes displayed more than 25 indels. Interestingly, 14 of the selected ATCC strains analyzed from public databases showed more than one assembly record, and three of these contained a different number of plasmids reported between the two separate assembles from the same strain identification (Table 3). Overall, we found that a considerable number of sequenced ATCC strains contain significant variations as compared to their public database counterparts. Without the accurate metadata and sample traceability, it is difficult to identify the source of the variation. In some cases, these variations may be attributable to the incorrect identification of the ATCC isolate before the sequence is submitted (eg, sequencing from a strain other than the intended ATCC strain). In other cases, the variations may have been caused by differences in strain propagation, DNA extraction, sequencing quality, or downstream assembly analysis, which could influence the overall quality of data in historical sequencing databases.
Table 3. Summary of variant call analysis for strains with more than one database record.
| Species | ATCC No. | Existing Reference Genomes | NCBI assembly level (plasmids*) | # of SNPs | # of indels | Average coverage (variants) |
|---|---|---|---|---|---|---|
| Acinetobacter baumannii | 17978 | GCA_001593425.2 | Complete genome | 14 | 5 | 210.1 |
| Acinetobacter baumannii | 17978 | GCA_000015425.1 | Complete genome (2) | 118 | 656 | 152.7 |
| Porphyromonas gingivalis | 33277 | GCA_000010505.1 | Complete genome | 20 | 7 | 319.5 |
| Porphyromonas gingivalis | 33277 | GCA_002892575.1 | Complete genome | 24 | 8 | 323.8 |
| Staphylococcus epidermidis | 12228 | GCA_002215535.1 | Complete genome (5) | 56,346 | 2,328 | 181.2 |
| Staphylococcus epidermidis | 12228 | GCA_000007645.1 | Complete genome (6) | 66 | 35 | 129.5 |
| Fusobacterium nucleatum | 25586 | GCA_003019295.1 | Complete genome | 29 | 14 | 310.4 |
| Fusobacterium nucleatum | 25586 | GCA_000007325.1 | Complete genome | 49 | 22 | 289.7 |
| Corynebacterium glutamicum | 13032 | GCA_000011325.1 | Complete genome | 18 | 2 | 216.7 |
| Corynebacterium glutamicum | 13032 | GCA_000196335.1 | Complete genome | 88 | 62 | 175.0 |
| Escherichia coli | 8739 | GCA_000019385.1 | Complete genome | 24 | 0 | 175.9 |
| Escherichia coli | 8739 | GCA_003591595.1 | Complete genome | 5 | 14 | 179.8 |
| Bifidobacterium longum | 15697 | GCA_000020425.1 | Complete genome | 14 | 7 | 336.1 |
| Bifidobacterium longum | 15697 | GCA_000269965.1 | Complete genome | 5 | 5 | 312.6 |
| Vibrio campbellii | BAA-1116 | GCA_000464435.1 | Complete genome [2 chr](1) | 198 | 336 | 143.0 |
| Vibrio campbellii | BAA-1116 | GCA_000017705.1 | Complete genome [2 chr](1) | 26 | 47 | 107.3 |
| Bacillus licheniformis | 14580 | GCA_000008425.1 | Complete genome | 17 | 4 | 174.4 |
| Bacillus licheniformis | 14580 | GCA_000011645.1 | Complete genome | 14 | 5 | 201.7 |
| Vibrio natriegens | 14048 | GCA_001456255.1 | Complete genome [2 chr] | 4 | 10 | 152.3 |
| Vibrio natriegens | 14048 | GCA_001680025.1 | Complete genome [2 chr] | 21 | 50 | 70.63 |
*Number in parentheses represent the number of plasmids reported in NCBI assembly report.
To support the sequence variation observed in ATCC genome sequences from public databases and assess the quality of our sequences, we performed independent short-read sequencing in duplicate using different experimental variables (Table 4). We then measured the reproducibility of our analysis via the number of SNVs and indels detected and the level of variant coverage observed.
Table 4. Summary of the reference base mapping analysis from multiple datasets.
| Test | Species | ATCC No. | Reference Genome | Analysis | # of SNPs | # of indels | Number of variants | Variant coverage |
|---|---|---|---|---|---|---|---|---|
| A | Mycoplasma hominis | 23114 | GCA_000085865.1 | Preparation 1 | 14 | 10 | 24 | 1042.1 |
| A | Mycoplasma hominis | 23114 | GCA_000085865.1 | Preparation 2 | 14 | 10 | 24 | 900.0 |
| A | Cutibacterium acnes | 11828 | GCA_000231215.1 | Preparation 1 | 28 | 37 | 65 | 121.1 |
| A | Cutibacterium acnes | 11828 | GCA_000231215.1 | Preparation 2 | 28 | 39 | 67 | 128.6 |
| B | Clostridium acetobutylicum | 824D-5 | GCA_000008765.1 | Kit 1 | 171 | 55 | 226 | 95.9 |
| B | Clostridium acetobutylicum | 824D-5 | GCA_000008765.1 | Kit 2 | 170 | 55 | 225 | 202.0 |
| B | Aeromonas hydrophila | 7966D-5 | GCA_000014805.1 | Kit 1 | 1 | 1 | 2 | 216.8 |
| B | Aeromonas hydrophila | 7966D-5 | GCA_000014805.1 | Kit 2 | 1 | 1 | 2 | 203.0 |
| C | Escherichia coli | 700926 | GCA_000005845.2 | Extraction 1 | 0 | 1 | 1 | 137.0 |
| C | Escherichia coli | 700926 | GCA_000005845.2 | Extraction 2 | 0 | 1 | 1 | |
| C | Streptococcus pyogenes | 19615 | GCA_000743015.1 | Extraction 1 | 2 | 44 | 46 | 314.2 |
| C | Streptococcus pyogenes | 19615 | GCA_000743015.1 | Extraction 2 | 2 | 41 | 43 | 460.2 |
Test A: Same DNA sequenced using two different DNA preparations
Test B: Same DNA sequenced with two different library kits
Test C: Same strain extracted with two different methods
Development and evaluation of hybrid de novo bacterial assembly
The results from the read alignment and mapping for variant detection in the ATCC published genomes demonstrated a diverse range of errors that impact the integrity of multiple reference genomes in public databases (Table 3). Therefore, it is essential that more sequence quality control and a standardized assembly approach are used to evaluate reference genome sequences.18 Several tools and NGS technologies used for de novo genome assembly have demonstrated the advantages of using a hybrid approach for the generation of precise genome sequences.19-21 With the goal of producing complete and circularized bacterial chromosomes and plasmids, and to provide a precise reference standard sequences from strains of our microbiology collection, we developed and standardized a hybrid de novo assembly and annotation bioinformatics pipeline. To minimize the potential effect of base call errors reported for ONT sequences,22 we developed a quality control protocol to trim and filter sequences by size and quality with fixed thresholds (Figure 4B).
Briefly, DNA from the ATCC collection and the extracted NGS-ready DNA were sequenced using both short-read and long-read NGS platforms. To continue the implementation of the best practices for our genome analysis workflow, and to reduce biases related to read length, sequence quality, coverage, and genome complexity like % GC and repeat regions, we standardized a method for sequencing DNA by using two NGS technologies and several instruments. First, this dual sequencing approach ensures the generation of high-quality contiguous and circular genome contigs with accurate base call and error polishing via high-quality Illumina short read coverage (median Q score, all bases > 30 and coverage threshold > 100x) and bacterial chromosome scaffolding and circularization with quality-filtered ultra-long reads obtained by Oxford Nanopore sequencing (minimum mean Q score, per reads >10, and minimum reads length > 5kb). Second, to guarantee a correct and complete de novo genome and to verify the taxonomic classification of the new reference ATCC genome sequences, we annotated and assessed the quality of genomes produced by our analysis pipeline by using previously published and certified bioinformatic tools (for additional details see ATCC Genome Portal Technical Document). Through this hybrid de novo assembly approach, we were able to generate complete circular chromosomes from ATCC certified strains, and we identified a diverse number of assembly errors (eg, single variants and chromosomal rearrangements) in the ATCC genome sequences from public databases (Figure 5, Table 5).
Table 5. Summary of metadata and quality of genome assemblies from public databases.
| ATCC No. | GenBank assembly | GenBank seq. platform | Year published to GenBank | # of variants between ATCC and GenBank assemblies | Average variant coverage | Structural variation detected |
|---|---|---|---|---|---|---|
| 8739 | GCA_000019385.1 | Not reported | 2008 | 20 | 159.9X | No |
| 12228 | GCA_002215535.1 | PacBio | 2017 | 58,674 | 181.2X | Yes |
| 13048 | GCA_003417445.1 | Ion Torrent | 2018 | 736 | 171.4X | n/a |
| 14028 | GCA_003864015.1 | PacBio | 2018 | 81 | 102.5X | Yes |
| 17978 | GCA_001593425.2 | Illumina MiSeq | 2016 | 21 | 204.0X | Yes |
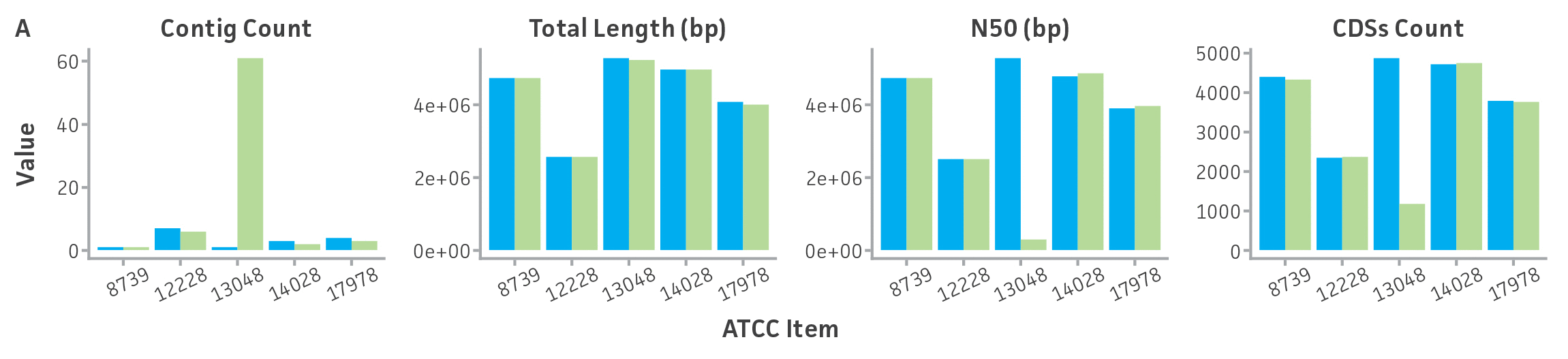
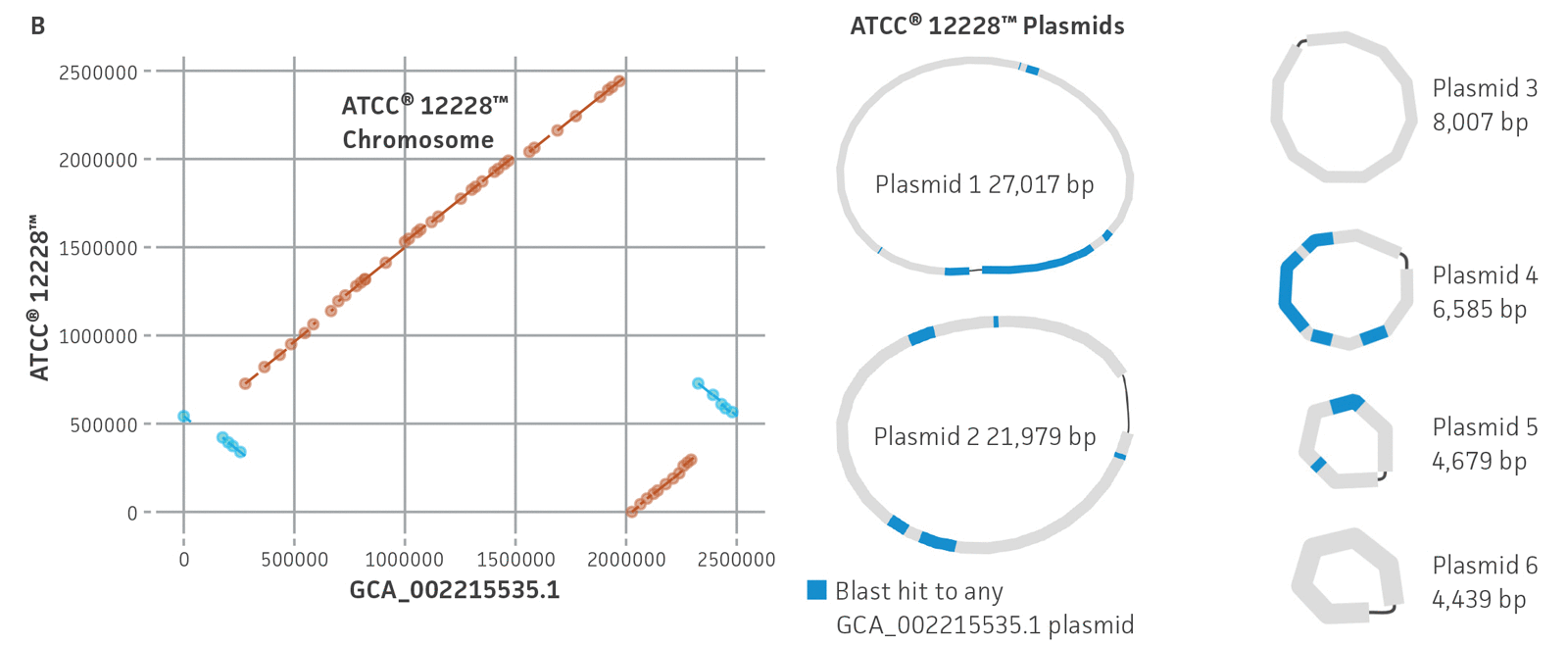
Figure 5. Evaluation of genome sequences from public databases. (A) Pairwise comparisons between select assembled ATCC genomes and their GenBank counterpart for a variety of assembly metrics. ATCC genomes (purple) show comparable or better assembly metrics than publicly available genomes (green). CDSs is coding sequences; N50 is the size of the shortest contig when 50% of the genome is contained in contigs of the same size or larger. (B) MUMmer24 alignment of ATCC de novo genome assembly of ATCC 12228 versus GenBank RefSeq genome assembly GCA_002215535.1, and plasmid alignments. Results are indicative of substantial structural variation and no complete matching plasmids between assemblies.
Quality control of genome assemblies
We used CheckM23 and taxonomic analysis to validate the quality of the assembly and the species designation (for additional details see ATCC Genome Portal Technical Document). Additionally, to support the quality of our analysis, we compared the ATCC genome assemblies developed using our workflow with the de novo hybrid assemblies (Nanopore and PacBio) produced by an external and certified third-party sequence facility that used same ATCC cultures. Here, twelve ATCC original cultures were extracted by an external laboratory and then sequenced and assembled using its analysis pipeline. To review the quality of the assemblies, we performed a WGS comparison between multiple datasets of the genome sequences obtained from our pipeline and the genome sequences assemblies obtained by the external lab (Figure 6, Table 6). Together, these results validate the reproducibility and confidence of our study and support the construction of an authenticated reference genome database.

Figure 6. Examples of whole-genome sequence alignments between ATCC assemblies and external source assemblies. (A) Whole-genome sequence alignments for Escherichia coli (ATCC 8739). (B) Whole-genome sequence alignments for Bacillus subtilis (ATCC 6633). Solid color blocks represent high sequence homology and structural similarity between two separated assemblies. Figures were generated using Mauve.25
Table 6. Summary of the de novo genome assemblies from multiple databases
| Species | ATCC No. | Sequence dataset | Total consensus (Mbp) | # of contigs (circular) | N50 (Mbp) | % GC |
|---|---|---|---|---|---|---|
| Bacillus subtilis | 6633 | Extraction 1 | 4.041 | 1 (1) | 4.041 | 43.9 |
| Bacillus subtilis | 6633 | Extraction 2 | 4.041 | 1 (1) | 4.041 | 43.9 |
| Bacillus subtilis | 6633 | Extraction 3 | 4.045 | 1 (1) | 4.045 | 43.9 |
| Bacillus subtilis | 6633 | Extraction 4* | 4.045 | 1 (1) | 4.045 | 43.9 |
| Escherichia coli | 8739 | Extraction 1 | 4.747 | 1 (1) | 4.747 | 50.9 |
| Escherichia coli | 8739 | Extraction 2 | 4.746 | 1 (1) | 4.746 | 50.9 |
| Escherichia coli | 8739 | Extraction 3* | 4.746 | 1 (1) | 4.746 | 50.9 |
| Staphylococcus aureus | 6538 | Extraction 1 | 2.800 | 2 (2) | 2.772 | 32.9 |
| Staphylococcus aureus | 6538 | Extraction 2 | 2.800 | 2 (2) | 2.772 | 32.9 |
| Staphylococcus aureus | 6538 | Extraction 3* | 2.800 | 2 (2) | 2.772 | 32.9 |
*Datasets obtained for DNA extraction, sequencing, and analysis from an external lab using ATCC Genuine Cultures
Finally, we evaluated several available annotation packages to select the most efficient, precise, and complete tool to provide the gene annotation for the final ATCC genome assemblies. Here, the genome assembly of Escherichia coli K-12 (ATCC 12435) was annotated using four different tools: Prokka, PGAP, EcoCyc (a combination of bioinformatics tools and manual curation by scientists), and AMG (an in-house advanced microbial genome annotation pipeline).26-28 In this analysis, we considered EcoCyc to be the “gold standard” for bacterial genome annotation. In general, the best results and most complete features were produced for PGAP (Table 7). The results suggest equivalent outputs of Prokka and PGAP and correct features specified for a gold standard annotation pipeline.
Table 7. Summary of features evaluated between genome annotation tools
| Feature | EcoCyc | PGAP | Prokka | AMG* |
|---|---|---|---|---|
| CDs | 4357 | 4377 | 4305 | 4325 |
| gene | 4566 | 4500 | 4416 | 4325 |
| misc_feature | 48 | 0 | 0 | 0 |
| misc_recomb | 1 | 0 | 0 | 0 |
| mobile_element | 49 | 0 | 0 | 0 |
| ncRNA | 72 | 13 | 0 | 0 |
| rRNA | 22 | 22 | 22 | 22 |
| regulatory | 0 | 0 | 0 | 0 |
| rep_origin | 1 | 0 | 0 | 0 |
| repeat_region | 697 | 2 | 0 | 0 |
| source | 1 | 1 | 1 | 1 |
| tRNA | 86 | 87 | 88 | 86 |
| tmRNA | 0 | 1 | 1 | 0 |
*AMGP: Advanced Microbial Genome Annotation pipeline developed by ATCC
Summary
As life science research progresses, the quality of data becomes increasingly important. Yet, the whole-genome sequencing data available in various public databases are frequently incomplete, fragmented, and contain errors. This is problematic as comprehensive, high-quality sequence data are essential for making correlations between in silico analyses and for translating research into clinical diagnostics and other regulated applications.
Therefore, as part of our initiative to enhance the authentication of biological materials, we have developed a standardized genome sequencing and assembly workflow to provide researchers with reference-grade genomes that are matched to authenticated ATCC strains. Our optimized methodology uses a hybrid assembly approach that combines the power of highly accurate Illumina short reads with the revolutionary scaffolding ability of Oxford Nanopore ultra-long reads. We have then taken our workflow one step further by accompanying each stage of the process with rigorous quality control analyses that ensure our data is the highest quality possible.
To date, ATCC’s ongoing genome sequencing efforts have produced over 250 authenticated reference-quality genomes for bacterial species pertinent to human health, diagnostics, quality control, microbiome studies, and antimicrobial resistance and rediscovery applications. We provide these sequences in a cloud-based portal that will enable researchers to quickly find and compare the data they need. We believe that this robust and high-quality ATCC genomic database will be of immense use to researchers for the development, verification, and validation of NGS-based assays in diverse areas of microbiology.
Download a PDF of this application note
Download Now
References
- Land M, et al. Insights from 20 years of bacterial genome sequencing. Funct Integr Genomics 15(2): 141-61, 2015.
- Deurenberg RH, et al. Application of next generation sequencing in clinical microbiology and infection prevention. J Biotechnol 243: 16-24, 2017.
- Swaminathan S, et al. Recent developments in genomics, bioinformatics and drug discovery to combat emerging drug-resistant tuberculosis. Tuberculosis (Edinb) 101: 31-40, 2016.
- Burgess DJ. Genomics: Next regeneration sequencing for reference genomes. Nat Rev Genet 19(3): 125, 2018.
- Zhulin IB. Databases for Microbiologists. J Bacteriol 197(15): 2458-67, 2015.
- Smits THM. The importance of genome sequence quality to microbial comparative genomics. BMC Genomics 20(1): 662, 2019.
- Sichtig H, et al. FDA-ARGOS is a database with public quality-controlled reference genomes for diagnostic use and regulatory science. Nat Commun 10(1): 3313, 2019.
- Tang L. Human gut bacterial genome reference. Nat Methods 16(4): 286, 2019.
- Uchiyama, I., et al., MBGD update 2018: microbial genome database based on hierarchical orthology relations covering closely related and distantly related comparisons. Nucleic Acids Res 47(D1): D382-D389, 2019.
- Balvociute M, Huson DH. SILVA, RDP, Greengenes, NCBI and OTT - how do these taxonomies compare? BMC Genomics 2017 Mar 14 [cited 18 Suppl 2]; 2017/04/01:[114]. Available from: https://www.ncbi.nlm.nih.gov/pubmed/28361695.
- Wattam AR, et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res 45(D1): D535-D542, 2017.
- EzBioCloud Database. EzBioCloud is ChunLab’s public data and analytics portal focusing on taxonomy, ecology, genomics, metagenomics, and microbiome of Bacteria and Archaea. Our new cloud service includes bioinformatics tools and succeeds our previous databases, which include EzTaxon, EzTaxon-e, and EzGenome. ]. Available from: https://www.ezbiocloud.net/.
- Joint Genome Institute (JGI). Genome Portal version:8.18.17 content:23b6a78052 jgi-portal-web-4.nersc.gov Release Date:16-Aug-2019 15:43:01.450 PST Current Date:05-Sep-2019 09:08:01.14 PDT]. Available from: https://genome.jgi.doe.gov/portal/.
- Field D, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 26(5): 541-7, 2008.
- Endrullat C, et al. Standardization and quality management in next-generation sequencing. Appl Transl Genome 10: 2-9, 2016.
- Tatusova P, et al. The NCBI Handbook: About Prokaryotic Genome Processing and Tools. 2nd edition ed. 2014.
- Criteria of the UniProt Knowledgebase. Available from: http//insideuniprot.blogspot.co.uk/2015_05_01_archive.html.
- Ma X, et al. Analysis of error profiles in deep next-generation sequencing data. Genome Biol. 20(1): 50, 2019.
- Sohn JI, Nam JW. The present and future of de novo whole-genome assembly. Brief Bioinform 19(1): 23-40, 2018.
- Miller JR, et al. Hybrid assembly with long and short reads improves discovery of gene family expansions. BMC Genomics 18(1): 541, 2017.
- Khan AR, et al. A Comprehensive Study of de novo Genome Assemblers: Current Challenges and Future Prospective. Evol Bioinform Online. 14: 1176934318758650, 2018.
- Laver T, et al. Assessing the performance of the Oxford Nanopore Technologies MinION. Biomol Detect Quantif 3: 1-8, 2015.
- Parks DH, et al. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25(7): 1043-55, 2015.
- Marcais G, et al. MUMmer4: A fast and versatile genome alignment system. PLOS Computational Biology 14(1): e1005944, 2018.
- Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5:e11147, 2010.
- Karp PD, et al. The EcoCyc Database. EcoSal Plus 8(1): doi: 10.1128/ecosalplus.ESP-0006-2018, 2018.
- Tatusova T, et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 44(14): 6614-6624, 2016.
- Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30(14): 2068-2069, 2014.
Acknowledgments
We would like to thank Nick Greenfield, MA, and Team at One Codex for their expertise and support with establishing the ATCC Genome Portal. Additionally, we thank Dev Mittar, PhD, for his innovation and leadership as well as Sam Minot, PhD, for his insight and helpful suggestions throughout the design and development process.