Why it’s important
Specific EUAs were issued in March, reducing the FDA review process and allowing for commercial distribution of emergency authorized products to combat the spread of COVID-19.3 The employment of emergency-use authorization was a critical step in the US government’s pandemic response, allowing for accelerated development and distribution of vaccines, therapeutic biologics, and numerous medical devices like surgical masks and other personal protective equipment (PPE), decontamination systems, ventilators, diagnostic tests, and more.
The program has been extremely successful. By the end of March 2020, there were already 21 authorized diagnostic assays on the market.3 That number would grow to 186 assays by the end of 2020 and 253 assays by the end of 2021. Given that the development cycle leading to 510(k) clearance of an in vitro diagnostic (IVD) assay can take up to two years, the speed at which assay developers were able to create their tests and receive authorization to commercialize them is truly astonishing.
What’s next
The EUAs to commercialize products will not go on forever. In December 2021, the FDA issued draft guidance for a transition back to the normal process of 510(k) submissions and premarket approval (PMA) guidelines.4 At the point of termination, emergency authorized products will no longer be eligible for commercial distribution unless they have received FDA clearance or approval or the developer has submitted all the required documentation to the FDA in order to achieve those designations. Manufacturers that choose not to pursue ongoing commercial distribution must also prepare as the FDA will expect proper labeling and customer notification of products with EUA versus products that are cleared or approved.How we can help
A critical detail of the transition plan is that only six months of notice will be given ahead of EUA termination—less time than would be needed to conduct the additional development work needed for a 510(k) filing or PMA submission. Any organization planning to maintain commercialization should start their assay validation work now. Development delays introduce risks for product recall, loss of revenue, and damage to reputations. ATCC can help as our cultures are widely cited in EUA submissions and 510(k) filings for in vitro diagnostic tests, and we provide products geared toward diagnostic development as they are packaged in quantitative formats that reduce handling time. Explore the available cultures and molecular standards for SARS-CoV-2 molecular diagnostics development.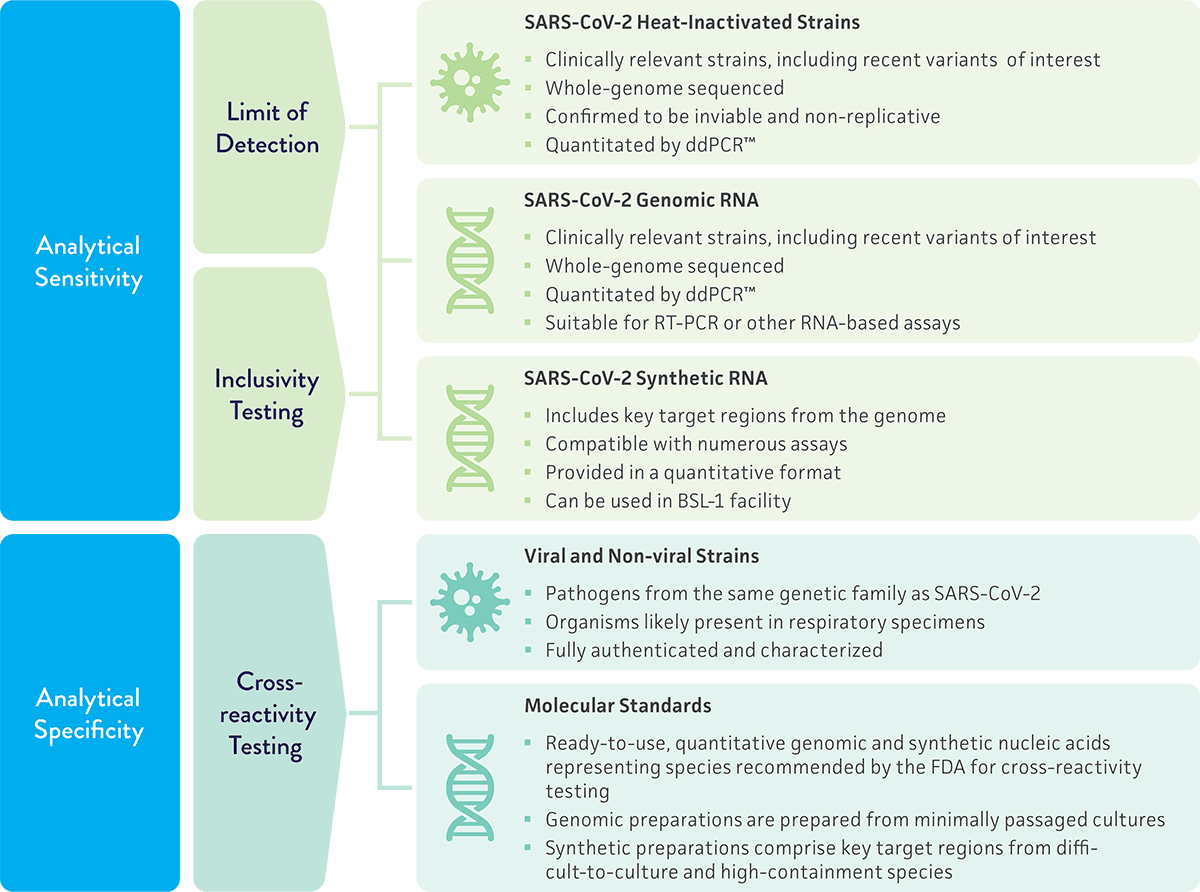
Kyle Young, MBA
Product Manager, ATCC
Kyle Young, MBA, is a Product Manager with 14 years of laboratory experience in virology and molecular biology. He has worked extensively with virus authentication at ATCC, leading several process development and improvement efforts. He has also been involved in the attainment of ISO certifications in several laboratories. He currently performs product line management work for ATCC’s Microbiology collections. Mr. Young earned a BS in Biology from the University of Tennessee and an MBA from George Mason University.
References
1. Public Health Emergency Declarations. https://www.phe.gov/emergency/news/healthactions/phe/Pages/default.aspx. Accessed 05May2022.
2. Emergency Use Authorization. https://www.fda.gov/emergency-preparedness-and-response/mcm-legal-regulatory-and-policy-framework/emergency-use-authorization#abouteuas. Accessed 05May2022.
3. Coronavirus Disease 2019 (COVID-19) Emergency Use Authorizations for Medical Devices, https://www.fda.gov/medical-devices/emergency-use-authorizations-medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices.
4. FDA. Draft Guidance for Industry and Food and Drug Administration Staff. Transition Plan for Medical Devices Issued Emergency Use Authorizations (EUAs) During the Coronavirus Disease 2019 (COVID-19) Public Health Emergency. Document issued December 2021. https://www.fda.gov/media/155039/download.