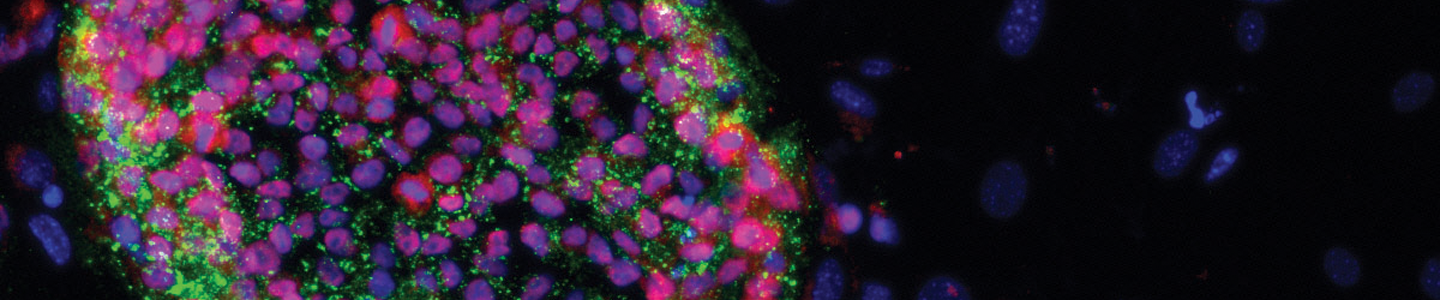
Stem cell culture remains a challenge, even with the knowledge that scientists have gained over the past decade. Learn best practices for keeping your stem cells happy.
Table of Contents
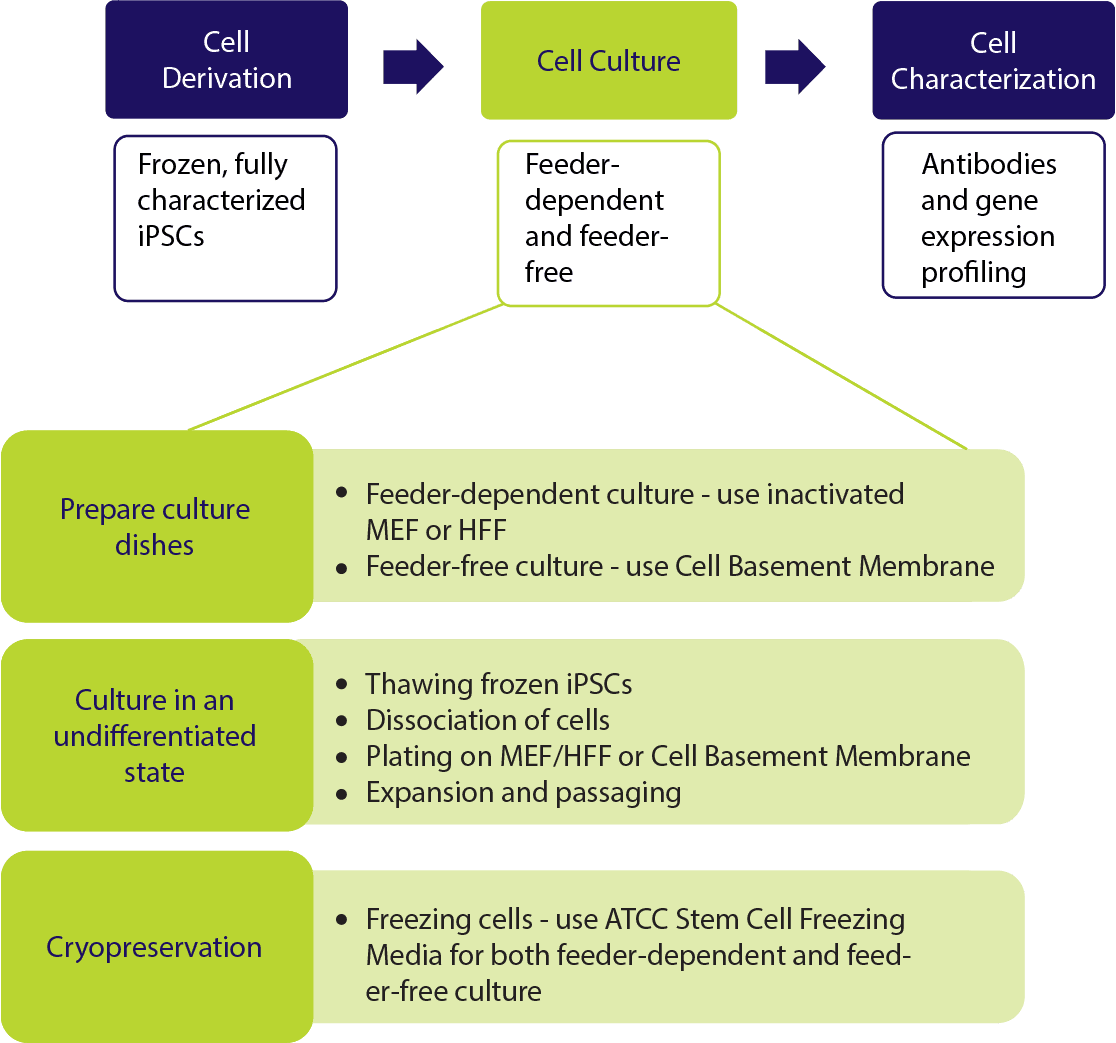
Stem Cell Culture Systems
ATCC Human Induced Pluripotent Stem Cell Culture
ATCC Mesenchymal Stem Cell Culture
ATCC Cancer Stem Cell Culture
Mouse Embryonic Stem Cell Culture Guide
Appendix
References
Get the ATCC guide to culturing stem cells
Download the GuideStem Cell Culture Systems
While stem cells hold great promise for biomedical research, the in vitro propagation and maintenance of these cells in an undifferentiated state is essential. Stem cell culture remains a challenge, even with the knowledge that scientists have gained over the past decade. Different types of stem cells often require different culture conditions. For example, human induced pluripotent stem cells (iPSC) and human embryonic stem cells (hESC) must be cultured on coated plates with a supporting layer of feeder cells, such as mouse embryonic fibroblasts (MEF) or human foreskin fibroblasts (HFF), or on an extracellular matrix, such as a basement membrane gel. In addition, stem cell growth media must be supplemented with growth factors. For example, fibroblast growth factor (FGF) should be added to mesenchymal stem cell (MSC) growth media and leukemia inhibitory factor (LIF) should be used when culturing mouse embryonic stem cells (mESC). Furthermore, some stem cells (such as iPSCs and hESCS) must have their media changed daily and monitored for overgrowth. Cell overgrowth will result in differentiation and lead to the loss of stem cell differentiative potential.
Table 1. Stem cells and selected culture conditions
| Stem Cell Types | Derived From | Attachment Support | Serum-free | ||
|---|---|---|---|---|---|
| Pluripotent | iPSCs | iPSCs | Post-natal tissues, such as skin, liver, heart | Yes (Feeder cells or extracellular matrix) | Yes |
| Pluripotent | ESCs | hESCs | blastocyst stage embryos | Yes (Feeder cells or extracellular matrix) | Yes |
| Pluripotent | ESCs | mESCs | blastocyst stage embryos | Yes (Feeder cells or gelatin) | No |
| Multipotent | Adult Stem Cells | MSCs | Post-natal tissues | No | No |
| Multipotent | Adult Stem Cells | Cancer Stem Cells (CSCs) | Tumors (such as brain, breast, colon) | No | Yes |
Feeder-dependent Stem Cell Cultures
A standard cell culture method utilized for maintaining iPSCs and mESCs is to co-culture with fibroblasts capable of conditioning the culture environment to support pluripotency and proliferative potentials of these stem cells. In feeder-dependent cell culture systems, the fibroblast-seeded plates need to be prepared in advance. Feeder cells condition the medium through metabolic leakage and provide a support matrix for cell attachment and proliferation. It is important to mitotically inactivate fibroblast-feeders so that they do not overgrow the stem cell culture. The feeder cells listed in the Appendix are either irradiated or treated with mitomycin C, which allows the feeder cells to continue to metabolize, but not to proliferate. Either MEFs or HFFs are recommended for iPSC cultures. The drawbacks with using feeders are 1) it can be more labor intensive, 2) it may pose a risk of transmitting animal pathogens to human stem cells if MEFs are used, and 3) it may yield mixed cultures of stem cells and fibroblasts. See the Appendix for a list of available ATCC feeder cell lines.
Feeder-free Stem Cell Cultures
Recent advances in stem cell culture technology have made it possible to culture pluripotent stem cells, such as iPSCs, in a feeder-free system by the utilization of extracellular matrices in the place of feeder cells. Like other pluripotent stem cells, iPSCs have a natural tendency to differentiate. Thus, the in vitro maintenance of undifferentiated iPSCs requires culture techniques that are distinct from those used with adult stem cells, continuous cell lines, or primary cells. The overall goal of feeder-free culture systems is to achieve a balance between promoting pluripotent cell growth and inhibiting spontaneous cellular differentiation by fine-tuning medium formulation of essential amino acids, salts, other nutritional elements and growth factors. Feeder-free culture conditions are 1) easier to use, 2) more reproducible, and 3) amenable to larger scales. In addition, the concern of obtaining mixed cultures of stem cells and fibroblasts is eliminated.
ATCC Stem Cell Solutions
ATCC has developed a growing portfolio of complete solution systems for successful stem cell culture and experiments. The ATCC stem cell collection, media, and reagents include:
- ATCC Human iPSCs: iPSCs derived from fibroblasts isolated from the skin, liver, and heart of normal tissue donors and donors with Parkinson’s disease, Down syndrome, or cystic fibrosis. These iPSCs have been reprogrammed by the episomal plasmid, retroviral, or Sendai viral expression of OCT4, SOX2, KLF4, and MYC genes. Also available: CD34+ iPSCs derived from bone marrow of normal tissue donors of various ethnicities and reprogrammed by Sendai viral expression.
- ATCC Human Mesenchymal Stem Cells: MSC lines isolated from single-donor umbilical cord, bone marrow, and adipose tissue and frozen at the second passage.
- ATCC Cancer Stem Cells: BT142 mut/- cell line isolated from an individual with grade III oligoastrocytoma containing the R132H mutation in isocitrate dehydrogenase I (IDH1).
- ATCC Mouse Embryonic Stem Cell Lines: mESC lines for producing transgenic mouse strains by homologous recombination and GFP/YFP expressing.
- ATCC Feeder Cells: These include regular and mitotically-inactivated mouse embryonic fibroblast and human foreskin fibroblast feeder cell lines.
- ATCC Stem Cell Media, Kits, and Reagents: These products include specially-formulated stem cell media and kits for growth, dissociation, cryopreservation, and differentiation.
See Appendix for a listing of ATCC Stem Cell Solutions products.
ATCC Human Induced Pluripotent Stem Cell Culture
Introduction
Human somatic cells can be genetically reprogrammed to become embryonic stem cell-like by introducing combinations of genes typically expressed during early embryogenesis. These reprogrammed cells are known as iPSCs.1 Questions remain, particularly about what clinical differences there are between iPSCs and ESCs. However, the versatility to create patient-specific iPSC lines and the ability to culture and differentiate them to a specific lineage gives the cells great potential for developmental and tissue differentiation studies, epigenetics, disease-modeling, drug development, and regenerative medicine.
iPSC Reprogramming Methods:
The introduction of the OCT4, SOX2, KLF4, and MYC genes (commonly known as the “Yamanaka factors”) is necessary to convert cells from post-natal tissues into iPSCs. Commonly employed methods for forced expression of these genes into cells are episomal plasmid, retroviral, and Sendai (RNA-based) virus. Each of these methods has its advantages and disadvantages, as described below:
- Episomal Expression: Recent improvements on this technique include the development of non-integrating episomes (high-expression plasmids). This technique has a lower reprogramming efficiency.2
- Retroviral Expression: This was the original method developed by Yamanaka and colleagues. While it offers the advantage of high reprogramming efficiency, the retroviral expression method leaves a genomic footprint.2
- Sendai Virus Expression: This technique offers the advantage of a high reprogramming efficiency and is not likely to leave a genomic footprint because the virus is non-integrating.2
ATCC Human Induced Pluripotent Stem Cell Lines
Substantial variation in the differentiation potential has been reported between iPSC lines.3 Many factors could contribute to the variation, such as the genetic background of the original material, reprogramming methods, genomic foot-prints, and culture conditions. Consequently, utilization of reference iPSC lines is important to iPSC-related research. ATCC offers a wide variety of frozen iPSCs (see Appendix) that are reprogrammed using all three reprogramming methods, including those derived from the skin, liver, heart, and bone marrow of normal tissue donors and donors with Parkinson’s disease, Down syndrome, and cystic fibrosis. ATCC iPSCs are well-characterized, robust, and are frozen at a low passage number. Further, ATCC iPSC lines have been fully characterized for the expression of pluripotent markers, differentiation potential for the three germ layers (ectoderm, endoderm, mesoderm), and karyotypic stability. These iPSC lines are optimized for use in feeder-free, serum-free culture, but they can be easily adapted for use in feeder- dependent cell culture systems.
Overview: Derivation, Culture, and Characterization of ATCC iPSCs
The following sections focus on maintenance, expansion, cryopreservation, and characterization of iPSCs. Cell culture protocols include serum-free and feeder-dependent, as well as serum-free and feeder-free systems.
Tips for Successful iPSC Culture
The protocols described in the subsequent sections should allow for the reproducible and trouble-free culture of iPSCs if they are followed correctly. The followings are some aspects of iPSC culture that the user should be aware of.
- Changing 100% of the cell culture medium daily.
- Monitoring cultures for morphological changes that indicate differentiation; less than 10% of iPSCs should exhibit a differentiated morphology.
- Taking care not to over-pipette the iPSCs more than 2 to 3 times during passaging; single cells will not establish colonies after seeding.
Additional tips are noted in blue boxes throughout this guide.
Feeder-free iPSC Cultures
In a feeder-free system, a biological matrix is used in place of fibroblast feeders to provide a surface for the attachment of iPSCs. Cell culture dishes are coated with Cell Basement Membrane, incubated for one hour and then are ready to use (Figure 1). ATCC iPSCs are cultured in the feeder-free system and therefore, no adaptation is required when starting your cultures.
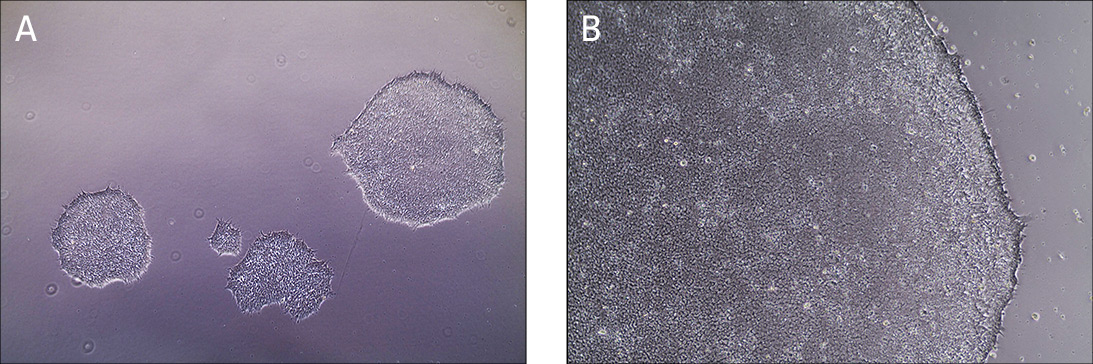
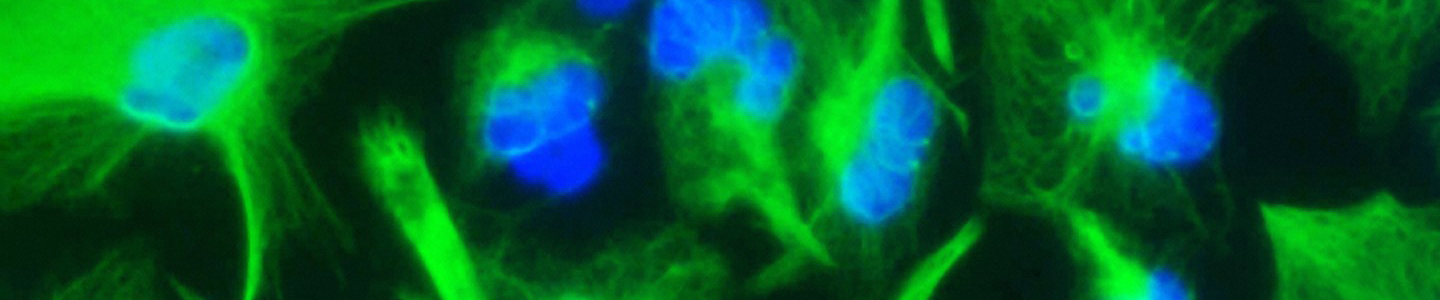
Figure 1. Characteristic morphology of iPSCs grown on feeder-free cultures using Pluripotent Stem Cell SFM XF/FF on Cell Basement Membrane.
(A) Undifferentiated iPSCs culture on Cell Basement Membrane in Pluripotent Stem Cell SFM XF/FF. (B) Undifferentiated iPSCs culture on Cell Basement Membrane in Pluripotent Stem Cell SFM XF/ FF. Note the round colony with tightly packed iPSCs with well-defined sharp edges. Individual cells within the colony exhibit prominent nucleoli and a high nucleus-to-cytoplasm volume ratio.
Materials for Feeder-free iPSC Culture
| ATCC No. | Product Name | Size | Storage |
|---|---|---|---|
| ACS-3002 | Pluripotent Stem Cell SFM XF/FF | 500 mL | -20°C |
| ACS-3010 | Stem Cell Dissociation Reagent | 250 mg | 2°C to 8°C |
| ACS-3030 | ROCK Inhibitor Y27632 | 10 mg | -20°C |
| ACS-3035 | Cell Basement Membrane | 5 mL | -80°C |
| 30-2006 | Dulbecco's Modified Eagle Medium (DMEM): F-12 Medium | 500 mL | 2°C to 8°C |
| 30-2200 | Dulbecco’s Phosphate Buffered Saline (D-PBS) | 500 mL | Room Temperature |
Preparation of Media Reagents
- Preparing Pluripotent Stem Cell SFM XF/FF complete medium
- SFM XF/FF is ready-to-use; no supplement is required. Thaw pluripotent Stem Cell SFM XF/ FF complete medium at 2°C to 8°C overnight. The thawed medium is stable for up to two weeks at 2°C to 8°C.
- Warm the medium for NOT MORE THAN 30 minutes in a 37°C water bath prior to use.
NOTE: It is recommended to prepare single use aliquots of the medium to prevent repeated warming to 37°C. If medium is not to be used within two weeks, it is acceptable to store aliquots of it at -20°C.
- Aliquoting Cell Basement Membrane for long-term storage
- Thaw Cell Basement Membrane on ice and swirl gently to mix. Important: Cell Basement Membrane will solidify in 15 to 30 minutes above 15°C. Keep Cell Basement Membrane, vials and pipette tips on ice at all times to prevent Cell Basement Membrane from solidifying. If air bubbles form, they may be eliminated by centrifuging Cell Basement Membrane at 300 x g for 10 minutes at 2°C to 8°C.
- Determine the appropriate volume per aliquot based on concentration and usage. The concentration of Cell Basement Membrane is found on the certificate of analysis.
Example: 2 mL of Cell Basement Membrane at 150 μg/mL is required to coat one 6-cm2 dish. To coat two 6-cm2 dishes, prepare as follows:
Dilute Cell Basement Membrane in DMEM:F12 to a working concentration of 150 μg/mL.
Protein concentration of Cell Basement Membrane (on certificate of analysis): 14 mg/mL:
(4 mL) x (0.15 mg/mL)/(14 mg/mL) = 0.043 mL
Add 43 μL Cell Basement Membrane directly in 4 mL cold DMEM: F-12 Medium.
Dispense appropriately-sized aliquots into pre-cooled tubes on ice and immediately place tubes at -20°C or -80°C for long-term storage.
NOTE: Product stability is extended by aliquoting the product in working volumes to avoid repeated freezing and thawing. Note that Cell Basement Membrane is diluted just prior to use. A protein concentration of 150 µg/mL is recommended for the propagation of stem cells.
- Reconstitution of Stem Cell Dissociation Reagent
Lyophilized proteins tend to be hygroscopic. Bring the vial of Stem Cell Dissociation Reagent to room temperature before opening. The vial should not be cool to the touch. Once opened, the lyophilized material should be stored desiccated. The specific activity of the reagent is found on the certificate of analysis. Prepare a 0.5 U/mL working solution and aliquot into working volumes according to usage.- Dissolve the appropriate amount of Stem Cell Dissociation Reagent in DMEM: F-12 Medium to prepare a 0.5 U/mL working solution.
Example: To prepare 40 mL of a 0.5 U/mL working solution:
Specific activity of Stem Cell Dissociation Reagent (on certificate of analysis) =1.46 U/mg (40 mL) x (0.5 U/mL)/(1.46 U/mg) = 13.7 mg
Dissolve 13.7 mg Stem Cell Dissociation Reagent in 40 mL DMEM: F-12 Medium. - Filter sterilize through a 0.22 µm filter membrane.
- Aliquot into working volumes according to routine usage.
- Store aliquots at -20°C for up to three months. Avoid repeated freezing and thawing. Thawed aliquots may be kept at 2°C to 8°C for up to two weeks.
- Dissolve the appropriate amount of Stem Cell Dissociation Reagent in DMEM: F-12 Medium to prepare a 0.5 U/mL working solution.
- Reconstitution of ROCK Inhibitor Y27632
ROCK Inhibitor Y27632 is soluble to 100 mM in water or D-PBS. We recommend preparation of a 10 mM stock solution as follows:- Add 3 mL of sterile water or D-PBS to the 10 mg vial of ROCK Inhibitor Y27632. Mix thoroughly.
- Aliquot into working volumes according to routine usage. Note that ROCK Inhibitor Y27632 is used at a final concentration of 10 μM (1:1000 dilution) in the cell culture medium.
- Store aliquots at -20°C. Avoid repeated freezing and thawing. Thawed aliquots may be kept at 2°C to 8°C for two weeks.
Prepare Cell Basement Membrane Coated Culture Dishes
This protocol is designed for coating two 6 cm2 dishes. Two (2) mL of Cell Basement Membrane is required per 6 cm2 dish. Volumes can be directly scaled according to the size and number of tissue culture vessels used.
Cell Basement Membrane will solidify in 15-30 minutes if the temperature is above 15°C. Keep Cell Basement Membrane and labware (pipette tips, serological pipettes, conical tubes) on ice at all times to prevent the matrix from gelling prematurely. If necessary, Cell Basement Membrane may be returned to a liquid state by placing it at 2°C to 8°C, on ice, for 24-48 hours.
- Remove one aliquot of Cell Basement Membrane from -20°C/-80°C storage and place on ice. Thaw Cell Basement Membrane in the refrigerator (2°C to 8°C), on ice, overnight.
- Sterilize the vial by rinsing with 70% ethanol. All of the operations from this point on should be carried out under strict aseptic conditions.
- Place 4 mL cold DMEM: F-12 Medium in a cold 15 mL conical tube on ice. Add the thawed Cell Basement Membrane to the 4 mL of DMEM: F-12 Medium on ice.
- Mix well. The final Cell Basement Membrane concentration should be 150 μg/mL (see previous section on aliquoting concentrated Cell Basement Membrane).
- Immediately coat the dishes with diluted Cell Basement Membrane. Use 2 mL for each 6 cm2 dish. Swirl dish gently to ensure that the entire dish is covered evenly.
NOTE: If air bubbles form when coating dishes, use a chilled pipette tip to break up the bubbles. - Leave coated dishes at 37°C for one hour.
- Aspirate coating solution and immediately plate the cells. It is critical that the coating does not dry out.
If the dishes will not be used the same day they are prepared, do not aspirate the coating solution. Seal the coated dishes with Parafilm and store at 2°C to 8°C for up to one week. Note that stored dishes should be warmed to room temperature in a biological safety cabinet for at least one hour before use.
Thawing of Cryopreserved iPSCs
- Pre-warm Pluripotent Stem Cell SFM XF/FF in a 37°C water bath. If using a small volume of medium (50 mL or less), warm only the volume needed in a sterile conical tube. Avoid warming complete medium multiple times.
NOTE: Prepare Pluripotent Stem Cell SFM XF/FF with ROCK Inhibitor by adding 20 μL of 10 mM ROCK Inhibitor Y27632 to 20 mL of medium. - Remove cryovial of frozen cells from liquid nitrogen storage.
- Thaw the cells by gently swirling in a 37°C water bath. To reduce the possibility of contamination, keep the O-ring and cap out of the water. Thawing should be rapid (approximately 1 to 2 minutes). Remove the cryovial from water bath when only a few ice crystals are remaining.
- Sterilize the cryovial by rinsing with 70% ethanol. All of the operations from this point on should be carried out under strict aseptic conditions.
- Use a 1 mL or 5 mL pipette to gently transfer cell suspension to a 15 mL conical tube.
- Slowly add 4 mL Pluripotent Stem Cell SFM XF/FF drop-wise, to the conical tube. Use an additional 1 mL of media to rinse the cryovial and transfer the liquid to the 15 mL tube. Shake the conical tube gently to mix the cells while adding media.
Do not break apart the aggregates into a single-cell suspension, as it is crucial to maintain the cells in clumps. - Centrifuge cells at 200 x g for 5 minutes at room temperature.
- Aspirate the supernatant and discard. Gently tap the bottom of the tube to loosen the cell pellet.
- Add 1 mL of stem cell culture medium with ROCK Inhibitor Y27632. Gently resuspend the pellet by pipetting up and down 2 to 3 times with a 1 mL tip. Take care not to over-pipette the culture into a single-cell suspension as single cells will not establish colonies after seeding.
NOTE: ROCK Inhibitor Y27632 is not necessary each time the culture medium is changed. It is recommended when the cells are recovering from thaw or being passaged.
Addition of ROCK inhibitor has been shown to increase the survival rate during subcultivation and thawing of human iPSCs. The use of ROCK inhibitor may cause a transient spindle-like morphology effect on the cells. However, the colony morphology will recover after subsequent media change without ROCK inhibitor. - Aspirate the coating solution from the plates prepared in the previous section (see Prepare Cell Basement Membrane Coated Culture Dishes). Add 4 mL of stem cell culture medium with ROCK Inhibitor Y27632 to each of two 6 cm2 dishes.
- Seed 0.5 mL of cell aggregates onto the dishes prepared in section Prepare Cell Basement Membrane Coated Culture Dishes.
- Incubate the culture at 37°C in a suitable incubator. A 5% CO2 in air atmosphere is recommended.
Maintenance of iPSC Cultures
Post thaw day 1, perform a 100% medium change and remove all cells that did not attach. Perform a 100% medium change every day.
- Changing Media
- Pre-warm Pluripotent Stem Cell SFM XF/FF in a 37°C water bath. If using a small volume of medium (50 mL or less), warm only the volume needed in a sterile conical tube. Avoid warming complete medium multiple times.
- Remove the cells from the incubator and view each dish under the microscope to determine percent cellular confluence and morphology of undifferentiated cells (see Maintenance of iPSC Cultures step 2 in Identification and Removal of Differentiated Cells). Characteristics of differentiation include colonies with less defined edges, dark areas, or non-uniform morphology.
- Carefully aspirate the medium without disturbing the monolayer.
- Add 4 mL of fresh, pre-warmed Pluripotent Stem Cell SFM XF/FF to the 6 cm2 dish and return the dish to the incubator.
- Every 24 hours, view each dish under the microscope to determine percent cellular confluence. If not ready to passage, repeat steps c and d as described above. When cells are cultured for 5 days or have reached approximately 80% confluence, with 90% undifferentiated cells, it is time to passage (see Passaging iPSCs).
It is essential that the cells are passaged before reaching confluence. Confluent colonies induce cell differentiation, along with exhibiting changes in morphology, slower proliferation and reduced differentiation capacity after passaging.
- Identification and Removal of Differentiated Cells
- Undifferentiated iPSCs grow as compact colonies and exhibit high nucleus-to-cytoplasm ratios and prominent nucleoli. The center of the cells will appear bright due to the multilayering of colonies. During the expansion and maintenance of iPSCs, however, differentiated cells may develop. Differentiating colonies have less-defined edges, dark areas, or exhibit a non-uniform morphology that is not typical of pluripotent stem cells such as fibroblast-like cells.
NOTE: We recommend less than 10% differentiation to maintain high quality cultures. Differentiated cells must be removed from the cell culture. - Observe cultures under the microscope for the appearance of differentiated cells (Figure 2). Use a lens marker to mark areas of differentiation on the dish.
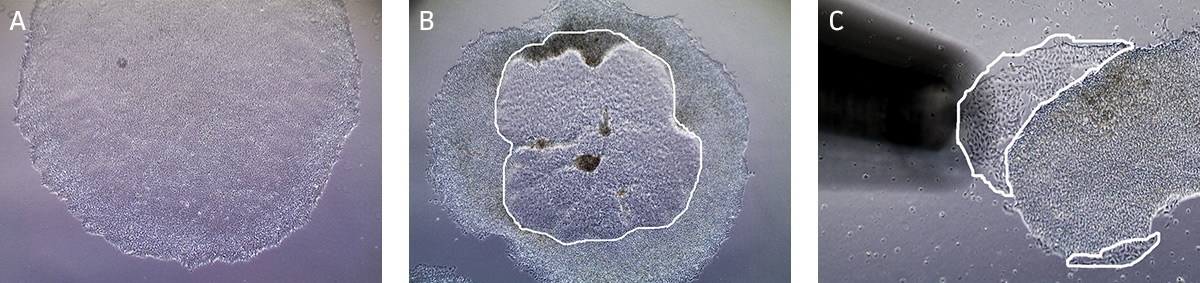
Figure 2. Characteristic morphology of iPSCs grown on feeder-free culture using Pluripotent Stem Cell SFM XF/FF on Cell Basement Membrane.
(A) Undifferentiated iPSC colony cultured in Pluripotent Stem Cell SFM XF/FF. (B) Partially differentiated iPSC colony cultured on Cell Basement Membrane. The circled region in the center of the colony designates the area of differentiated iPSCs that needs to be removed. (C) Partially differentiated iPSCs (circled) being removed/aspirated using a 200 µL pipette tip attached to the end of the aspirating pipette. - To remove clusters of differentiated cells, attach a fine-tipped aspirating pipette to a vacuum source. The tip can be made smaller by attaching a 200 μL pipette tip to the end of the aspirating pipette. Suction away portions of colonies which appear differentiated, as marked in step b. Take care not to allow the cultures to dry out.
- Undifferentiated iPSCs grow as compact colonies and exhibit high nucleus-to-cytoplasm ratios and prominent nucleoli. The center of the cells will appear bright due to the multilayering of colonies. During the expansion and maintenance of iPSCs, however, differentiated cells may develop. Differentiating colonies have less-defined edges, dark areas, or exhibit a non-uniform morphology that is not typical of pluripotent stem cells such as fibroblast-like cells.
- When to Passage Cells
Cells are typically split at a 1:4 ratio when cells are cultured for 5 days or 80% confluent (Figure 3). When colonies have grown such that adjacent colonies merge, or differentiation occurs in the center of each colony, the colonies are ready for passage.
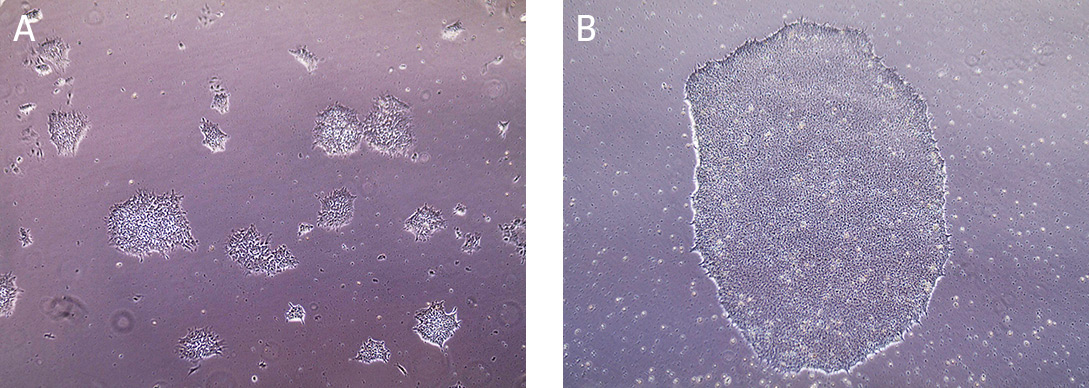
Figure 3. Morphology of iPSCs cultured on Cell Basement Membrane.
(A) Undifferentiated iPSCs 2 days post passage. (B) Undifferentiated iPSCs 5 days post passage, ready to split.
Passaging iPSCs
The protocol is designed for the dissociation and expansion of cells on a 6 cm2 dish. Volumes should be adjusted according to the size of the tissue culture vessels used.
NOTE: Make certain culture dishes coated with Cell Basement Membrane are prepared; see section on Cell Basement Membrane.
- Remove an aliquot of 0.5 U/mL Stem Cell Dissociation Reagent working solution from the freezer and allow it to warm to room temperature (15°C to 25°C). Please see Preparation of Media Reagents, Step 3, for instructions on how to prepare Stem Cell Dissociation Reagent.
- Pre-warm Pluripotent Stem Cell SFM XF/FF in a 37°C water bath. If using a small volume of medium (50 mL or less), warm only the volume needed in a sterile conical tube. Avoid warming complete medium multiple times. NOTE: Addition of ROCK inhibitor has been shown to increase the survival rate during subcultivation and thawing of human iPSCs. The use of ROCK inhibitor may cause a transient spindle-like morphology effect on the cells. However, the colony morphology will recover after subsequent media change without ROCK inhibitor.
- Aspirate the medium from the cells.
- Rinse cells twice by adding and removing 4 mL/dish of D-PBS.
- Add 2 mL of Stem Cell Dissociation Reagent working solution to each dish.
- Incubate at 37°C for 10 to 15 minutes. The reaction is complete when the edges of the individual colonies begin to loosen and fold back from the dish. View the plate under the microscope starting at 5 minutes as incubation time may vary depending on the cell line being used and colony size.
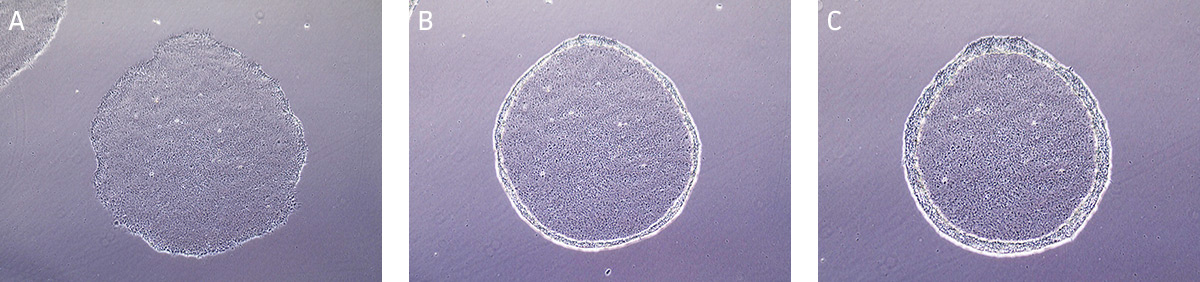
Figure 4. Time lapse images of passaging hiPSCs cultured on Cell Basement Membrane using Stem Cell Dissociation Reagent.
(A) Undifferentiated iPSC colony ready for passaging. (B) iPSC colony after 5 minutes in Stem Cell Dissociation Reagent, edges of the iPSC colony is starting to lift/curl. (C) iPSC colony after 10 minutes in Stem Cell Dissociation Reagent, ready to passage. - Carefully aspirate the Stem Cell Dissociation Reagent and gently rinse the colonies by adding and removing 4 mL/ dish of DMEM: F-12 Medium, taking care not to dislodge the cells during manipulation.
NOTE: Stem Cell Dissociation Reagent must be completely removed from the cells since the reagent is not inactivated by medium or serum. - Add 2 mL of Pluripotent Stem Cell SFM XF/FF with ROCK Inhibitor Y27632 to the dish, and detach the cells by pipetting up and down 2 to 3 times with a 1-mL tip. Take care not to over-pipette the culture into a single-cell suspension as single cells will not establish colonies after seeding. If cells are not detaching, use a cell scraper to detach the cells.
- Transfer the cell aggregates to a 15 mL conical tube.
NOTE: Prepare Pluripotent Stem Cell SFM XF/FF with ROCK Inhibitor by adding 20 µL of 10 mm ROCK Inhibitor Y27632 to 20 mL of medium. - Add an additional 3 mL of Pluripotent Stem Cell SFM XF/FF with ROCK Inhibitor Y27632 to the surface of the dish to collect any remaining cells. Transfer the rinse to the 15 mL conical tube containing the cell aggregates.
- Centrifuge cells at 200 x g for 5 minutes at room temperature to pellet cells.
- Aspirate the supernatant and discard.
- Add 1 mL of Pluripotent Stem Cell SFM XF/FF with ROCK inhibitor Y27632. Gently re-suspend the pellet by pipetting up and down 2 to 3 times with a 1 mL tip, maintaining the small cell aggregates. Take care not to over-pipette the culture into a single-cell suspension as single cells will not establish colonies after seeding.
- Plate the cells as desired on feeder or feeder-free cultures.
- Incubate the culture at 37°C in a humidified 5% CO2/95% air incubator. Perform a 100% medium change every day. Passage the cells every 4 to 5 days (80% confluent).
Feeder-dependent iPSC Culture
In feeder-dependent cell culture systems, the fibroblast-seeded plates need to be prepared in advance. iPSCs are easily distinguishable from the fibroblasts as the colonies are well defined and compact. Individual iPSCs within the colony are tightly packed and have a high nucleus-to-cytoplasm volume ratio (Figure 5).
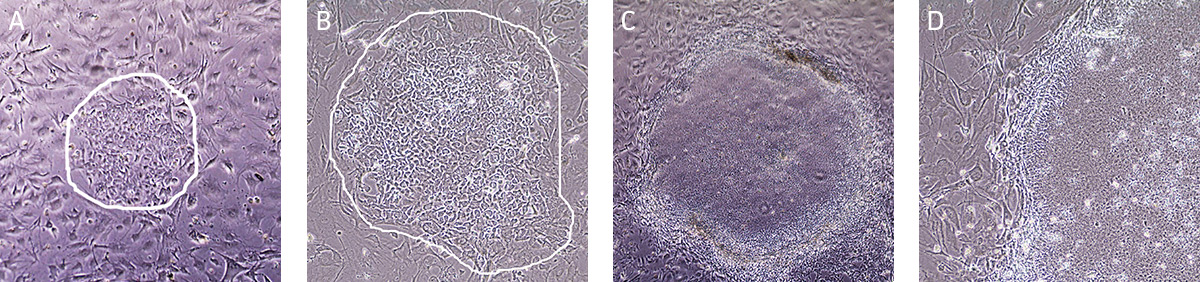
Figure 5. Characteristic morphology of hiPSCs grown on MEFs using Pluripotent Stem Cell SFM XF.
(A) A small iPSC colony (circled) with loose colony morphology. (B) A small iPSC colony (circled) with loose colony morphology. (C) Undifferentiated iPSC colony ready to passage with tightly packed iPSCs with well-defined sharp edges. (D) Individual cells within the colony exhibit prominent nucleoli with high nucleus-to-cytoplasm volume ratio.
Materials for Feeder-dependent iPSC Culture
| ATCC No. | Product Name | Size | Storage |
|---|---|---|---|
| ACS-3002 | Pluripotent Stem Cell SFM XF/FF | 500 mL | -20°C |
| ACS-3030 | ROCK Inhibitor Y27632 | 10 mg | -20°C |
| 30-2002 | DMEM | 500 mL | 2°C to 8°C |
| 30-2006 | DMEM: F-12 Medium | 500 mL | 2°C to 8°C |
| 30-2020 | Fetal Bovine Serum (FBS) | 500 mL | -20°C |
| 30-2200 | D-PBS | 500 mL | Room Temperature |
Preparation of Media Reagents
- Pluripotent Stem Cell SFM XF/FF:
Pluripotent Stem Cell SFM XF/FF medium is ready to use; no supplement is required. However, if 500 mL of medium will not be consumed within two weeks, thaw and aliquot the medium into desired volumes (eg, 100 mL) and store at -20°C. - DMEM + 15% FBS (Feeder medium):
- Thaw FBS at room temperature.
- Add FBS to a final concentration of 15% in DMEM.
- Complete medium can be stored at 2°C to 8°C for up to two weeks.
- Reconstitution of Stem Cell Dissociation Reagent:
Prepare a 0.5 U/mL working solution in DMEM: F-12 Medium and aliquot into working volumes according to usage. See Preparation of Media Reagents step 3, Reconstitution of Stem Cell Dissociation Reagent, in Feeder-free iPSC Culture section, for details. - Reconstitution of ROCK Inhibitor Y27632:
Prepare a 10 mM stock solution in D-PBS. See Preparation of Media Reagents step 4 for details.
Preparation of Feeder Cell Coated Dishes
Important: Cells should be plated 24 hours before use as a feeder layer and kept for no more than seven days.
- Pre-warm DMEM + 15% FBS (Feeder Medium) in a 37°C water bath. If using a small volume of medium (50 mL or less), warm only the volume needed in a sterile conical tube. Avoid warming complete medium multiple times.
- Pipet 5 mL of DMEM + 15% FBS to a 15 mL conical tube.
- Thaw one vial of MEF or HFF by gently swirling in a 37°C water. To reduce the possibility of contamination, keep the O-ring and cap out of the water. Thawing should be rapid (approximately 1 to 2 minutes). Remove the cryovial from water bath when a few ice crystals remain.
- Sterilize the vial by rinsing with 70% ethanol. All of the operations from this point on should be carried out under strict aseptic conditions.
- Use a 1 mL or 5 mL pipette to gently transfer cell suspension to a 15 mL conical tube containing DMEM + 15% FBS. Use an additional 1 mL of medium to rinse the vial and transfer the contents to the 15 mL tube.
- Add 4 mL of DMEM + 15% FBS, bringing the total volume to 10 mL.
- Gently mix and pellet the cells by centrifugation at 200 x g for 5 minutes.
- Discard the supernatant and resuspend the cells with 10 mL of fresh, pre-warmed DMEM + 15% FBS.
- Count viable cells (eg, using erythrosin B exclusion). Cell viability should be > 85%.
NOTE: Optimal feeder cell density is critical to maintain typical iPSC morphology. - Add the appropriate amount of fresh, pre-warmed DMEM + 15% FBS to plate cells at a seed density of 1.2 x 104 cells/cm2. Refer to Table 2 for suggested plating volumes.
- Incubate the feeder cells at 37°C and 5% CO2. Cells should be plated 24 hours before use and stored at 37°C and 5% CO2 for no more than 7 days.
When storing plates, DMEM + 15% FBS should be changed twice during the week or when pH decreases. It is important to avoid excessive alkalinity of the medium during the recovery of the cells.
Table 2. MEFs/HFFs Seeding Densities for Different Culture Dishes (Calculated with 12,000 cells/cm2)
| Cell Culture Vessel | Growth Area (cm2) | Total Number of Feeder Cells per Dish | Optimal Volume for Plating (mL) |
|---|---|---|---|
| 24 well | 2.0 | 2.4 x 104 | 0.5 |
| 12 well | 4.0 | 4.8 x 104 | 1.0 |
| 6 well / 35 mm2 | 9.5 | 1.2 x 105 | 2.0 |
| 6 cm2 | 21 | 3.4 x 105 | 4.0 |
| 10 cm2 | 56 | 9.4 x 105 | 10 |
Thawing of Cryopreserved iPSCs
- Pre-warm Pluripotent Stem Cell SFM XF/FF in a 37°C water bath. If using a small volume of medium (50 mL or less), warm only the volume needed in a sterile conical tube. Avoid warming complete medium multiple times.
NOTE: Before thawing cells, make certain culture dishes seeded with MEF or HFF are prepared. See Preparation of Feeder Cell Coated Dishes. - Replace the MEF or HFF feeder dish medium (DMEM + 15% FBS) with 4 mL of Pluripotent Stem Cell SFM XF/FF in the presence of 10 μM ROCK Inhibitor Y27632. Place dish in the incubator for 15 minutes to allow the medium to reach its normal pH (7.0-7.6). Two 6 cm2 plates are needed for each vial of cells thawed.
- Remove cryovial of frozen cells from liquid nitrogen storage.
- Thaw the cells by gently swirling in a 37°C water bath. To reduce the possibility of contamination, keep the O-ring and cap out of the water. Thawing should be rapid (approximately 1 to 2 minutes). Remove the cryovial from water bath when only a few ice crystals are remaining.
- Sterilize the cryovial with 70% ethanol. All of the operations from this point on should be carried out under strict aseptic conditions.
- Use a 1 mL or 5 mL pipette to gently transfer cell suspension to a 15 mL conical tube.
- Slowly add 4 mL Pluripotent Stem Cell SFM XF/FF, drop-wise, to the conical tube. Use an additional 1 mL of media to rinse the cryovial and transfer the liquid to the 15 mL tube. Shake the conical tube gently to mix the cells while adding media.
- Gently pipette the cells up and down 2 or 3 times to mix thoroughly. Note: Do not break apart the aggregates into a single-cell suspension, as it is crucial to maintain the cells in clumps.
- Centrifuge cells at 200 x g for 5 minutes at room temperature.
- Aspirate the supernatant and discard. Gently tap the bottom of the tube to loosen the cell pellet.
- Add 1 mL of Pluripotent Stem Cell SFM XF/FF in the presence of 10 μM ROCK Inhibitor Y27632 to the tube. Gently resuspend the pellet by pipetting up and down 2 or 3 times with a 1 mL tip, maintain the cell aggregates. Note: Do not break apart the aggregates into a single-cell suspension, as it is crucial to maintain the cells in clumps.
- Seed 0.5 mL of cell aggregates onto two 6 cm2 MEF or HFF feeder dishes, as prepared in step 2.
- Change medium the next day and daily thereafter until the cells have been cultured for 5 days or the colonies reach 80% confluency. ROCK Inhibitor Y27632 is not required in subsequent cell culture medium changes.
Maintenance of Feeder-dependent iPSC Cultures
- Changing Media
- Pre-warm Pluripotent Stem Cell SFM XF/FF in a 37°C water bath. If using a small volume of medium (50 mL or less), warm only the volume needed in a sterile conical tube. Avoid warming medium multiple times.
NOTE: Characteristics of differentiation include colonies with less defined edges, dark areas, or non-uniform morphology. - Remove the cells from the incubator and view each dish under the microscope to determine percent cellular confluence and morphology of undifferentiated cells. (See Maintenance of Feeder-dependent iPSC Cultures step 2, Identification and Removal of Differentiated Cells in Maintenance of iPSC Culture.)
- Carefully aspirate the medium without disturbing the monolayer.
- Add 4 mL of fresh, pre-warmed Pluripotent Stem Cell SFM XF/FF to the 6 cm2 dish and return the dish to the incubator.
- Every 24 hours, view each dish under the microscope to determine percent cellular confluence. If the cells are not ready to passage, repeat steps c and d as described above. When cells are cultured for 5 days or cultures have reached approximately 80% confluence, with 90% undifferentiated cells, it is time to passage.
- Pre-warm Pluripotent Stem Cell SFM XF/FF in a 37°C water bath. If using a small volume of medium (50 mL or less), warm only the volume needed in a sterile conical tube. Avoid warming medium multiple times.
- Identification and Removal of Differentiated Cells
We recommend less than 10% differentiation to maintain high quality cultures. Differentiated cells must be removed from the cell culture.- Undifferentiated iPSCs grow as compact colonies and exhibit high nucleus-to-cytoplasm ratios and prominent nucleoli. When cultured in Pluripotent Stem Cell SFM XF/FF, small iPSC colonies may initially exhibit loose colony morphology but will become more compact once the colony grows bigger. In addition, the colonies maintain a distinct border on feeder cells. During the expansion and maintenance of iPSCs, however, differentiated cells or other cell types may develop. Differentiating colonies have less defined edges, dark areas, or exhibit a non-uniform morphology that is not typical of pluripotent stem cells such as fibroblast-like cells (Figure 6).
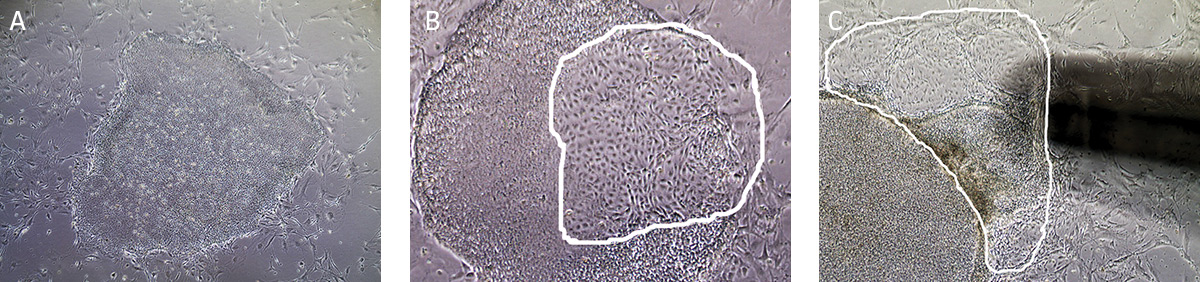
Figure 6. Characteristic morphology of iPSCs grown in feeder-dependent culture on MEFs.
(A) Undifferentiated iPSC colony cultured on MEFs. (B) Partially differentiated iPSC colony cultured on MEFs. The circled region designates the area of differentiated iPSCs that needs to be removed. (C) Partially differentiated iPSCs (circled) being removed/aspirated using a 200 µL pipette tip attached to the end of the aspirating pipette. - Observe cultures under the microscope for the appearance of differentiated cells. Use a lens marker to mark areas of differentiation on the dish.
- To remove clusters of differentiated cells, attach a fine-tipped aspirating pipette to a vacuum source. The tip can be made smaller by attaching a 200 μL pipette tip to the end of the aspirating pipette. Suction away portions of colonies which appear differentiated, as marked in step a. Take care not to allow the cultures to dry out.
- Undifferentiated iPSCs grow as compact colonies and exhibit high nucleus-to-cytoplasm ratios and prominent nucleoli. When cultured in Pluripotent Stem Cell SFM XF/FF, small iPSC colonies may initially exhibit loose colony morphology but will become more compact once the colony grows bigger. In addition, the colonies maintain a distinct border on feeder cells. During the expansion and maintenance of iPSCs, however, differentiated cells or other cell types may develop. Differentiating colonies have less defined edges, dark areas, or exhibit a non-uniform morphology that is not typical of pluripotent stem cells such as fibroblast-like cells (Figure 6).
- When to Passage Cells
iPSC cultures are typically split at a 1:4 ratio when cells are 80% confluent, after approximately 4 to 5 days. When colonies have grown such that adjacent colonies merge, or differentiation occurs in the center of each colony, the colonies are ready for passage. (Figure 7).
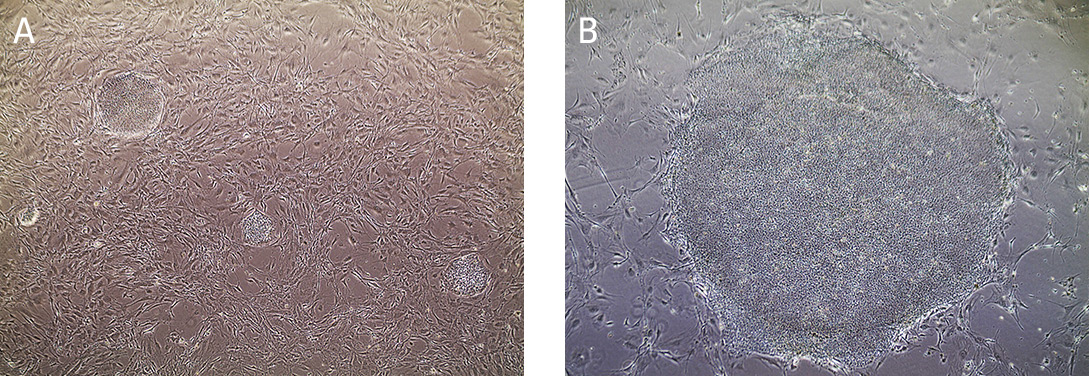
Figure 7. Morphology of hiPSCs cultured on MEF feeders.
(A) Undifferentiated iPSCs 2 days post passage. (B) Undifferentiated iPSCs 5 days post passage, ready to split.
Passaging Feeder-dependent iPSC Cultures
The protocol is designed for the dissociation and expansion of cells on a 6 cm2 dish. Volumes should be adjusted according to the size and number of the tissue culture vessels used.
NOTE: Make certain culture dishes coated with feeder cells are prepared. See Preparation of Dishes with Feeder Cells.
- Remove an aliquot of 0.5 U/mL Stem Cell Dissociation Reagent working solution from the freezer and allow it to warm to room temperature.
- Pre-warm Pluripotent Stem Cell SFM XF/FF in a 37°C water bath. If using a small volume of medium (50 mL or less), warm only the volume needed in a sterile conical tube. Avoid warming complete medium multiple times.
- Aspirate and discard the stem cell culture medium.
- Rinse cells twice by adding and removing 4 mL/dish of D-PBS.
- Add 2 mL of Stem Cell Dissociation Reagent working solution to each dish.
- Incubate at 37°C for 10-15 minutes. The reaction is complete when the edges of the individual colonies begin to loosen and fold back from the dish (Figure 8). View the plate under the microscope starting at 5 minutes as incubation time may vary depending on the cell line and colony size.
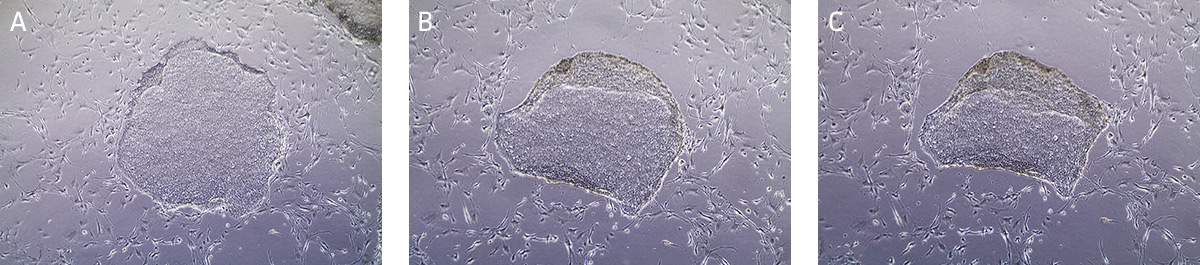
Figure 8. Time lapsed images of passaging hiPSCs cultured on MEFs using Stem Cell Dissociation Reagent.
(A) Undifferentiated iPSC colony ready for passaging. (B) iPSC colony after 5 minutes in dissociation reagent, edges of the iPSC colony is starting to lift/curl. (C) iPSC colony after 13 minutes in dissociation reagent, ready to passage. - Carefully aspirate the Stem Cell Dissociation Reagent and gently rinse the colonies by adding and removing 4 mL/dish of DMEM: F-12 Medium, taking care not to dislodge the cells during manipulation.
- Add 2 mL of Pluripotent Stem Cell SFM XF/FF to each dish. Detach cells by pipetting up and down 2 to 3 times with a 1 mL tip. Note: Do not break apart the aggregates into a single-cell suspension, as it is crucial to maintain the cells in clumps.
- Transfer the cell aggregates to a 15 mL conical tube.
- Add an additional 4 mL of Pluripotent Stem Cell SFM XF/FF to collect any remaining cells on the surface of the dish. Transfer rinse to the 15 mL conical tube containing the cell aggregates.
- Centrifuge cells at 200 x g for 5 minutes at room temperature to pellet cells.
- Aspirate the supernatant. Gently tap the bottom of the tube to loosen the cell pellet.
- For each dish processed, add 2 mL of Pluripotent Stem Cell SFM XF/FF in the presence of 10 μM ROCK Inhibitor Y27632 to the 15 mL conical tube.
NOTE: Prepare Pluripotent Stem Cell SFM XF with ROCK inhibitor by adding 20 μL of 10 mM ROCK Inhibitor Y27632 to 20 mL of medium. - Gently resuspend the pellet by pipetting up and down 2 to 3 times with a 1 mL tip, maintaining the small cell aggregates. Note: Do not break apart the aggregates into a single-cell suspension, as it is crucial to maintain the cells in clumps.
- If desired, fibroblasts can be depleted from cultures as follows:
- Add the entire suspension to an uncoated 6 cm2 tissue culture dish with 4 mL of Pluripotent Stem Cell SFM XF/FF. Incubate for one hour at 37°C, 5% CO2. During this time, the fibroblasts will adhere while the iPSCs clusters remain in suspension.
- Collect the medium into a 15 mL conical tube and centrifuge at 200 x g for 5 minutes at room temperature to pellet cells.
- Aspirate the supernatant. Gently tap the bottom of the tube to loosen the cell pellet.
- For each dish processed, add 2 mL of Pluripotent Stem Cell SFM XF/FF in the presence of 10 μM ROCK Inhibitor Y27632 to the 15 mL conical tube.
- Transfer 0.5 mL of the cell aggregates onto MEF-or HFF-coated dishes that contain 4 mL Pluripotent Stem Cell SFM XF/FF in the presence of 10 μM ROCK Inhibitor Y27632.
- Swiftly move the dishes in a forward to backward, then left to right pattern, once, to gently disperse the cells evenly across the surface of the dishes. Incubate dishes overnight at 37°C and 5% CO2.
- Change medium daily until the colonies are big enough to passage. ROCK Inhibitor Y27632 is not required in subsequent cell culture medium changes.
Cryopreservation of iPSCs
Materials:
| ATCC No. | Product Name | Size | Storage |
|---|---|---|---|
| ACS-3002 | Pluripotent Stem Cell SFM XF/FF | 500 mL | -20°C |
| ACS-3010 | Stem Cell Dissociation Reagent | 250 mg | 2°C to 8°C |
| ACS-3020 | Stem Cell Freezing Media | 20 mL | 2°C to 8°C |
| 30-2006 | DMEM: F-12 Medium | 500 mL | 2°C to 8°C |
| 30-2200 | D-PBS | 500 mL | Room Temperature |
Protocol for Cryopreservation
- Remove an aliquot of 0.5 U/mL Stem Cell Dissociation Reagent working solution from the freezer and allow it to warm to room temperature.
- Aspirate and discard the stem cell medium.
- Rinse the cells twice by adding and removing 4 mL/6 cm2 dish of D-PBS.
NOTE: For optimal results, cells should be cultured to 80% confluency before freezing. - Add 2 mL of Stem Cell Dissociation Reagent working solution to each dish.
- Incubate at 37°C for 10-15 minutes or until the edges of the individual colonies begin to loosen and fold back. View the dish under the microscope starting at 5 minutes as incubation time may vary depending on the cell line and colony size.
- Aspirate the Stem Cell Dissociation Reagent and gently rinse the colonies by adding and removing 4 mL/dish of DMEM:F-12 Medium, taking care not to dislodge the cells during manipulation.
- Add 2 mL of stem cell culture medium to each dish and detach cells by pipetting up and down 2 to 3 times with a 1 mL tip. Take care not to over-pipette the culture into a single-cell suspension. It is best if the cells remain aggregated.
NOTE: One 6 cm2 dish yields 2 mL of frozen cells. Cells are frozen in 1 mL aliquots. - Transfer the cell aggregates to a 15 mL conical tube.
- Add an additional 3 mL of stem cell culture medium to the dish to collect any remaining cells. Transfer this rinse to the 15 mL conical tube containing the cell aggregates.
- Centrifuge cells at 200 x g for 5 minutes at room temperature to pellet cells. While cells are spinning, remove the Stem Cell Freezing Media from storage and swirl to mix. Keep cold. Decontaminate by dipping in or spraying with 70% alcohol.
- Aspirate the supernatant and discard. Gently tap the bottom of the tube to loosen the cell pellet.
- Add 2 mL of cold Stem Cell Freezing Media to the tube. Gently resuspend the pellet by pipetting up and down 2 to 3 times with a 1 mL tip, maintaining the small cell aggregates. Cells will recover from the freezing/thawing process more effectively if cells are frozen in clumps.
- Immediately transfer 1 mL each of the cell suspension into two cryovials.
- Freeze the cells gradually at a rate of -1°C/min until the temperature reaches -70°C to -80°C. A cryopreservation container (eg, CoolCell freezing container, ATCC ACS-6000) may also be used.
- The cells should not be left at -80°C for more than 24 to 48 hours. Once at -80°C, frozen cryovials should be transferred to the vapor phase of liquid nitrogen for long-term storage.
Characterization of iPSCs
Morphology
Undifferentiated iPSCs grow as compact colonies and exhibit high nucleus-to-cytoplasm ratios and prominent nucleoli (Figures 9A and 9B). When cultured in Pluripotent Stem Cell SFM XF/FF medium, small iPSC colonies may initially exhibit loose colony morphology but will become more compact as the colony grows larger. In addition, the iPSC colonies maintain a distinct border on feeder cells (Figure 9C). During the expansion and maintenance of iPSCs, differentiated cells or obvious alternate cell types might emerge (Figure 9D, arrow). These cells should be removed before passaging the cultures by attaching a 200 μL pipette tip to the end of the aspirating Pasteur pipette cells (see Maintenance of iPSC Cultures step 2 in Identification and Removal of Differentiated Cells).
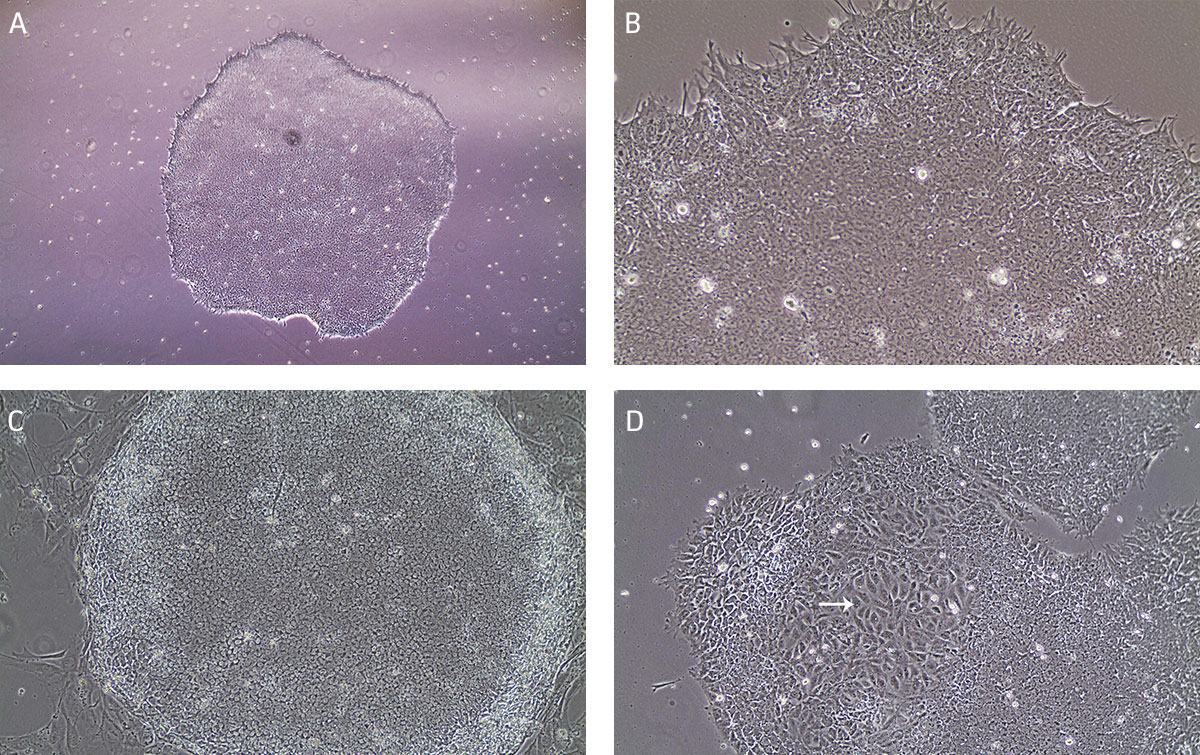
Figure 9. Characteristic morphology of iPSCs grown on feeder-free or feeder-dependent culture.
(A) Undifferentiated iPSC colony cultured on Cell Basement Membrane in Pluripotent Stem Cell SFM XF/FF grows as a compact colony with individual cells displaying high nucleus-to -cytoplasm ratios and prominent nucleoli. (B) Undifferentiated iPSC colony cultured on Cell Basement Membrane in Pluripotent Stem Cell SFM XF/FF at high magnification. (C) Undifferentiated iPSC colony cultured on MEF in Pluripotent Stem Cell SFM XF/FF. Note the defined border on feeder cells. (D) Arrow indicates partially differentiated iPSCs grown on feeder-free culture. Differentiated colonies exhibit less defined edges and non-uniform morphology.
Characterization Assays
- Immunocytochemistry
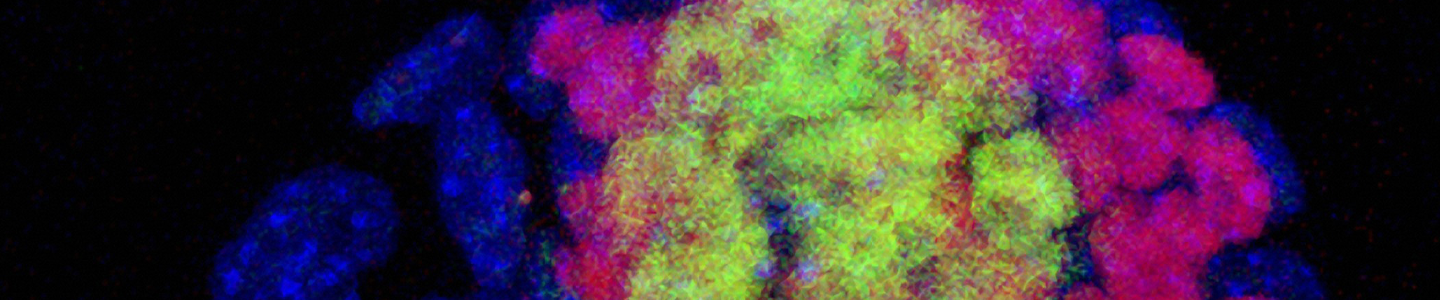
Immunocytochemistry is the most accessible and easiest method for most laboratories to assess the pluripotency of iPSCs cultures. The cells are fixed and then incubated with primary antibodies to pluripotency markers. These antibodies are then detected with a secondary fluorophore-conjugated antibody. Undifferentiated iPSCs can be characterized by the expression of pluripotent markers such as SSEA4, Tra-1-60, Tra-1-81, and Nanog (Figure 10).
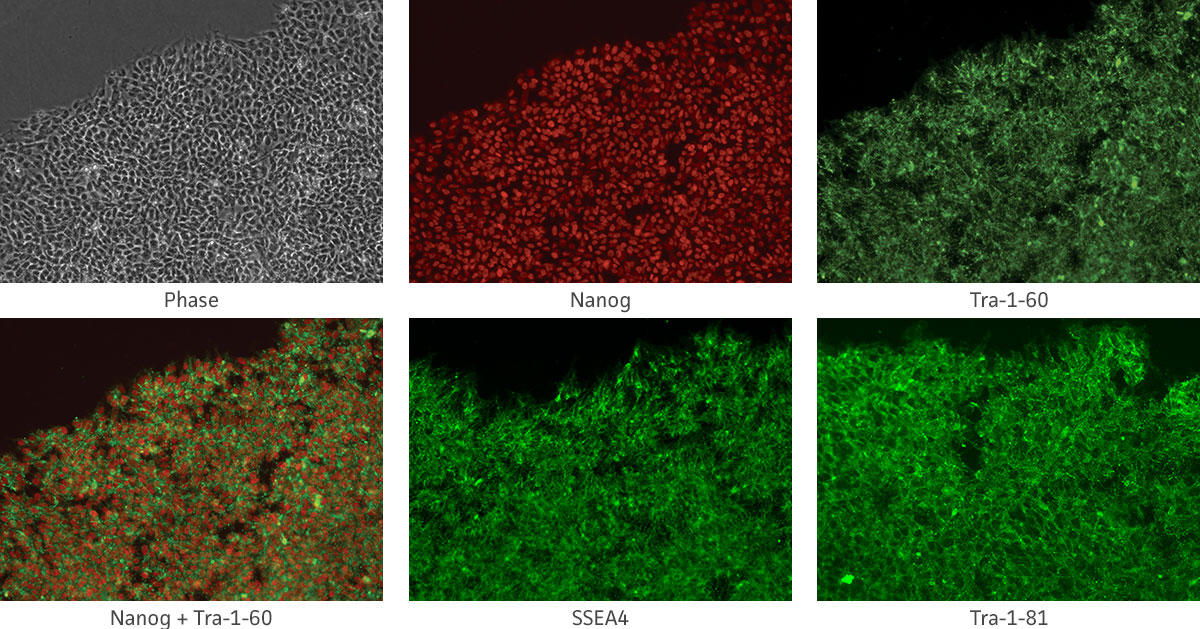
Figure 10. Immunocytochemistry of iPSCs stained with Tra 1-60, Tra-I-81, SSEA4, and Nanog antibodies (10x).
- Flow Cytometry
To quantitate the percentage of undifferentiated iPSCs in culture, flow cytometric analysis of pluripotency markers and differentiation markers is performed. Samples of cells are stained with a fluorophore conjugated antibody to the pluripotency surface markers of interest. The cells are then counted by a flow cytometer. Upon differentiation of iPSCs, Tra-1-60 and SSEA4 expression levels decrease while SSEA1 expression increases. For a pure, undifferentiated iPSC line, the expression level of SSEA4 and Tra-1-60 pluripotency markers should be more than 90% of the total cells and the expression level of SSEA1 should be less than 15% of the total cells (Figure 11).
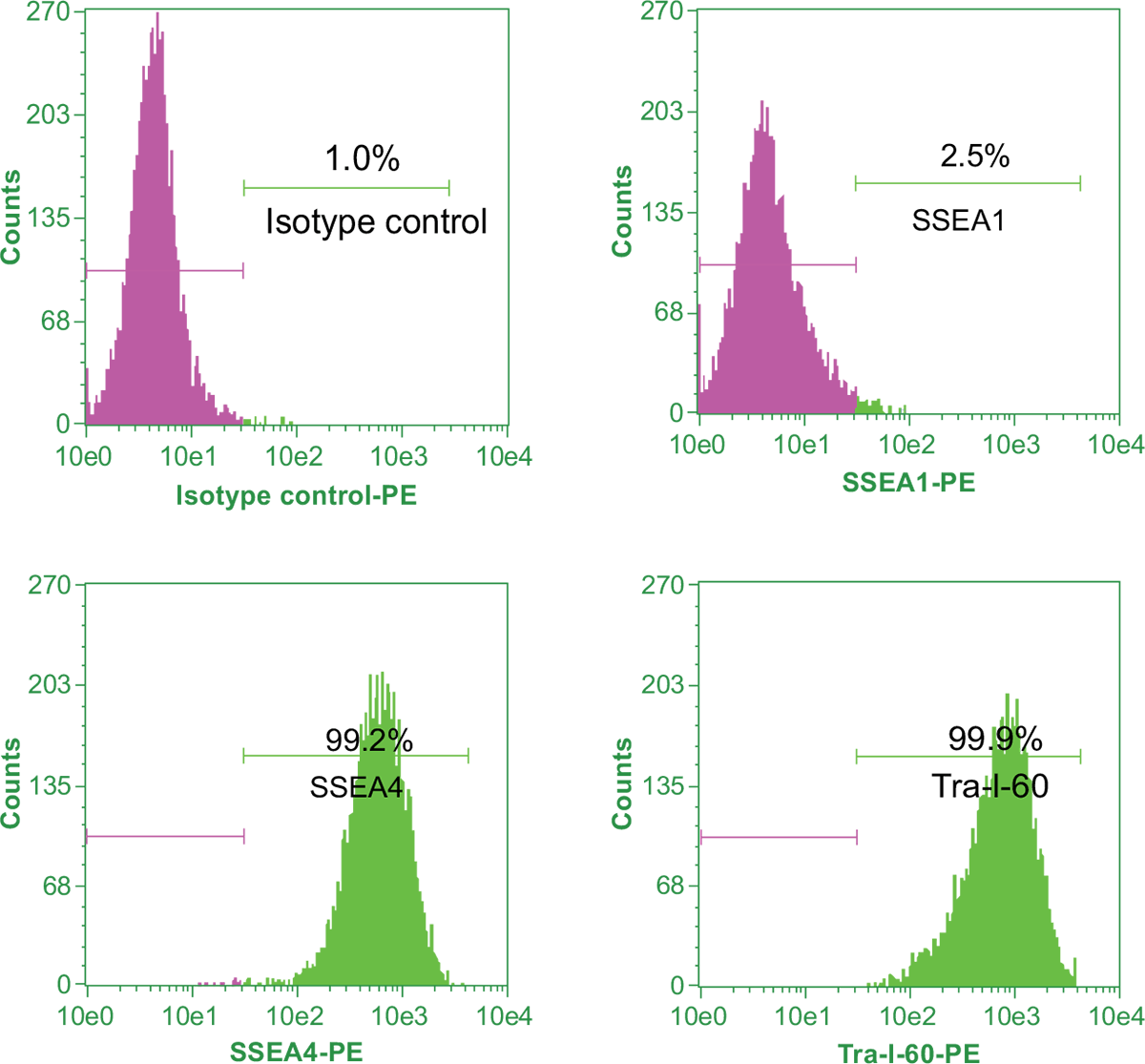
Figure 11. Flow cytometric analysis of expression of pluripotency markers in iPSCs.
- Karyotype
Stem cells can develop karyotypic mutations over time. It is good practice to verify that the iPSCs are karyotypically normal after several passages. Various methods are available including comparative genomic hybridization array, Giemsa banding, and fluorescent in situ hybridization analysis. - STR Analysis
Short Tandem Repeat (STR) testing is a rapid, reproducible PCR-based technique used for the identification or authentication of iPSC lines.
Characterization Markers and Techniques
Table 3. Markers Commonly Used for Characterizing Pluripotent Stem Cells
| Marker | Subcellular Location | iPSC expression |
|---|---|---|
| Alkaline Phosphatase | Intracellular | Upregulated in pluripotent cells |
| Tra-1-60 | Cell surface | Upregulated in pluripotent cells |
| Tra-1-81 | Cell surface | Upregulated in pluripotent cells |
| SSEA-1 | Cell surface | Downregulated in pluripotent cells |
| SSEA-4 | Cell surface | Upregulated in pluripotent cells |
| Nanog | Nuclear | Upregulated in pluripotent cells |
Table 4. Comparison of Standard Methods Used for Characterizing Pluripotent Stem Cells
| Method | Application | Comments |
|---|---|---|
| Live stain | For selection of pluripotent colonies after reprogramming | Cells remain viable, allowing for continued expansion. Use when images from a limited number of markers is adequate. |
| Immunocytochemistry | Characterization of one or multiple markers of pluripotency | Cells are fixed but can test for a broad range of markers. Excellent when vivid images are desired and loss of cell sample is not an issue. |
| Flow cytometry | Characterization of one or multiple markers of pluripotency | Relative quantitation of percentage of pluripotent versus differentiated cells in a culture. |
ATCC Mesenchymal Stem Cell Culture
Introduction
MSCs are self-renewing, multipotent adult stem cells. MSCs are isolated from the connective tissues throughout the body, such as in the bone marrow4, adipose, human umbilical cord or cord blood, and peripheral blood. These precursor cells can be differentiated into bone, fat, and cartilage in vitro upon treatment with certain factors.5
MSCs are useful in understanding cell differentiation as well as tissue engineering6-9, orthopedic and obesity research, regenerative medicine10-13, and the creation of iPSC cell lines. Lipoaspirates and umbilical cord tissue represent a heterogeneous mixture of cell types, including adipocytes, endothelial cells, smooth muscle cells, pericytes, and progenitor cells. The heterogeneity of the source material increases the potential that the MSC culture will be contaminated and potentially over-run by another cell type. As a result, consistent isolation of MSCs from such material can be challenging, is time consuming, and is sometimes less cost-effective.
ATCC Mesenchymal Stem Cell Solutions
ATCC offers four types of human mesenchymal stem cells: Umbilical Cord-derived Mesenchymal Stem Cells (ATCC PCS-500-010), Adipose-derived Mesenchymal Stem Cells (ATCC PCS-500-011), Bone Marrow-derived Mesenchymal Stem Cells (ATCC PCS-500-012), and hTERT Immortalized Adipose-derived Mesenchymal Stem Cells (ASC52telo, ATCC SCRC-4000). Adipose-derived MSCs are isolated from human adipose (fat) tissues by lipoaspiration or biopsy. MSCs derived from the umbilical cord are isolated from Wharton’s Jelly, the gelatinous substance within the human umbilical cord. Bone marrow-derived MSCs are isolated from bone marrow aspirates. In addition, all four cell types are isolated from single-donor tissue. ATCC MSCs are cryopreserved at second passage and tested for growth, morphology, marker expression and differentiation potential. ATCC also offers mesenchymal cell growth media, reagents, and differentiation kits. These are described in the following tables as well as on our website www.atcc.org/stemcells.
Mesenchymal Stem Cell Culture Protocols
Materials
Table 5. Mesenchymal Stem Cells and Complete Growth Medium
| Mesenchymal Stem Cells | Growth Kit Options | Basal Medium |
|---|---|---|
| Adipose-derived Mesenchymal Stem Cells, Normal, Human (ATCC PCS-500-011) | Mesenchymal Stem Cell Growth Kit for Adipose- and Umbilical-derived MSCs - Low Serum (ATCC PCS-500-040) | Mesenchymal Stem Cell Basal Medium (ATCC PCS-500-030) |
| Umbilical Cord-derived Mesenchymal Stem Cells; Normal, Human (ATCC PCS-500-010) | Mesenchymal Stem Cell Growth Kit for Adipose- and Umbilical-derived MSCs - Low Serum (ATCC PCS-500-040) | Mesenchymal Stem Cell Basal Medium (ATCC PCS-500-030) |
| ASC52telo, hTERT immortalized Adipose-derived Mesenchymal Stem Cells (ATCC SCRC-4000) | Mesenchymal Stem Cell Growth Kit for Adipose- and Umbilical-derived MSCs - Low Serum (ATCC PCS-500-040) | Mesenchymal Stem Cell Basal Medium (ATCC PCS-500-030) |
| Bone Marrow-derived Mesenchymal Stem Cells; Normal, Human (ATCC PCS-500-012) | Mesenchymal Stem Cell Growth Kit for Bone Marrow-derived MSCs (ATCC PCS-500-041) | Mesenchymal Stem Cell Basal Medium (ATCC PCS-500-030) |
| Reagents for Subculture | ||
| D-PBS (ATCC 30-2200) | ||
| Trypsin-EDTA for Primary Cells (ATCC PCS-999-003) | ||
| Trypsin Neutralizing Solution (ATCC PCS-999-004) | ||
Preparation of Media Reagents
Note: For Bone Marrow-derived MSCs refer to the website at www.atcc.org/stemcells.
- Obtain one Mesenchymal Stem Cell Growth Kit–Low Serum from the freezer; make sure that the caps of all components are secure.
- Thaw the components of the growth kit just prior to adding them to the basal medium.
- Obtain one bottle of Mesenchymal Stem Cell Basal Medium from cold storage.
- Decontaminate the external surfaces of all growth kit component vials and the basal medium bottle by spraying them with 70% ethanol.
- Using aseptic technique and working in a laminar flow hood or biosafety cabinet, transfer the indicated volume of each growth kit component, according to the following table, to the bottle of basal medium using a separate sterile pipette for each transfer.
Table 6. Mesenchymal Stem Cell Growth Kit for Adipose and Umbilical-derived MSCs - Low Serum (ATCC PCS-500-040)
| Component | Volume | Final Concentration |
|---|---|---|
| MSC Supplement | 10 mL | 2% FBS 5 ng/mL rh FGF basic 5 ng/mL rh FGF acidic 5 ng/mL rh epidermal growth factor (EGF) |
| L-Alanyl-L-Glutamine | 6 mL | 2.4 mM |
- Tightly cap the bottle of complete growth medium and swirl the contents gently to assure a homogeneous solution. Do not shake forcefully to avoid foaming. Label and date the bottle.
- Complete growth media should be stored in the dark at 2°C to 8°C (do not freeze). When stored under these conditions, complete growth media is stable for two weeks.
Note: Refer to the batch specific information provided on the Certificate of Analysis for the total number of viable cells recovered from each lot of ATCC Human MSCs.
Thawing of Cryopreserved MSCs
- Using the total number of viable cells, determine how much surface area can be inoculated to achieve an initial seeding density of 5,000 cells per cm2.
- Prepare the desired combination of flasks. Add 5 mL of complete growth medium per 25 cm2 of surface area. Place the flasks in a 37°C, 5% CO2, humidified incubator and allow the media to pre-equilibrate to temperature and pH for 30 minutes prior to adding cells.
- While the culture flasks equilibrate, remove one vial of ATCC Human MSCs from storage and thaw the cells by gentle agitation in a 37°C water bath. To reduce the possibility of contamination, keep the O-ring and cap out of the water. Thawing should be rapid (approximately 1 to 2 minutes).
- Remove the vial from the water bath as soon as the contents are thawed, and decontaminate by dipping in or spraying with 70% ethanol. All operations from this point onward should be carried out under strict aseptic conditions.
- Add the appropriate volume of complete growth medium [volume = (1 mL x number of flasks to be seeded) – 1 mL] into a sterile conical tube. Using a sterile pipette, transfer the cells from the cryovial to the conical tube. Gently pipette the cells to homogenize the suspension. Do not centrifuge.
- Transfer 1 mL of the cell suspension to each of the pre-equilibrated culture flasks prepared in steps 1 to 3 of Handling Procedure for Frozen Cells and Initiation of Culture. Pipette up and down several times, then cap and gently rock each flask to distribute the cells evenly.
- Place the seeded culture flasks in a 37°C, 5% CO2, incubator. Incubate for at least 24 hours before processing the cells further.
Maintenance
- Pre-warm the complete growth media in a 37°C water bath. This will take between 10 and 30 minutes, depending on the volume. If using a small volume of medium (50 mL or less), warm only the volume needed in a sterile conical tube. Avoid warming complete growth media multiple times.
- Remove the cells from the incubator 24 hours after seeding, and then view each flask under the microscope to determine percent cellular confluence.
- Remove the spent media carefully without disturbing the monolayer.
- Add 5 mL of fresh, pre-warmed complete growth medium per 25 cm2 of surface area and return the flasks to the incubator.
- View each flask under the microscope 24 to 48 hours after seeding to determine the percent cellular confluence. If not ready to passage, repeat steps 3 and 4 as described above. When cells have reached approximately 70% to 80% confluence, and are actively proliferating (many mitotic figures are visible), it is time to subculture.
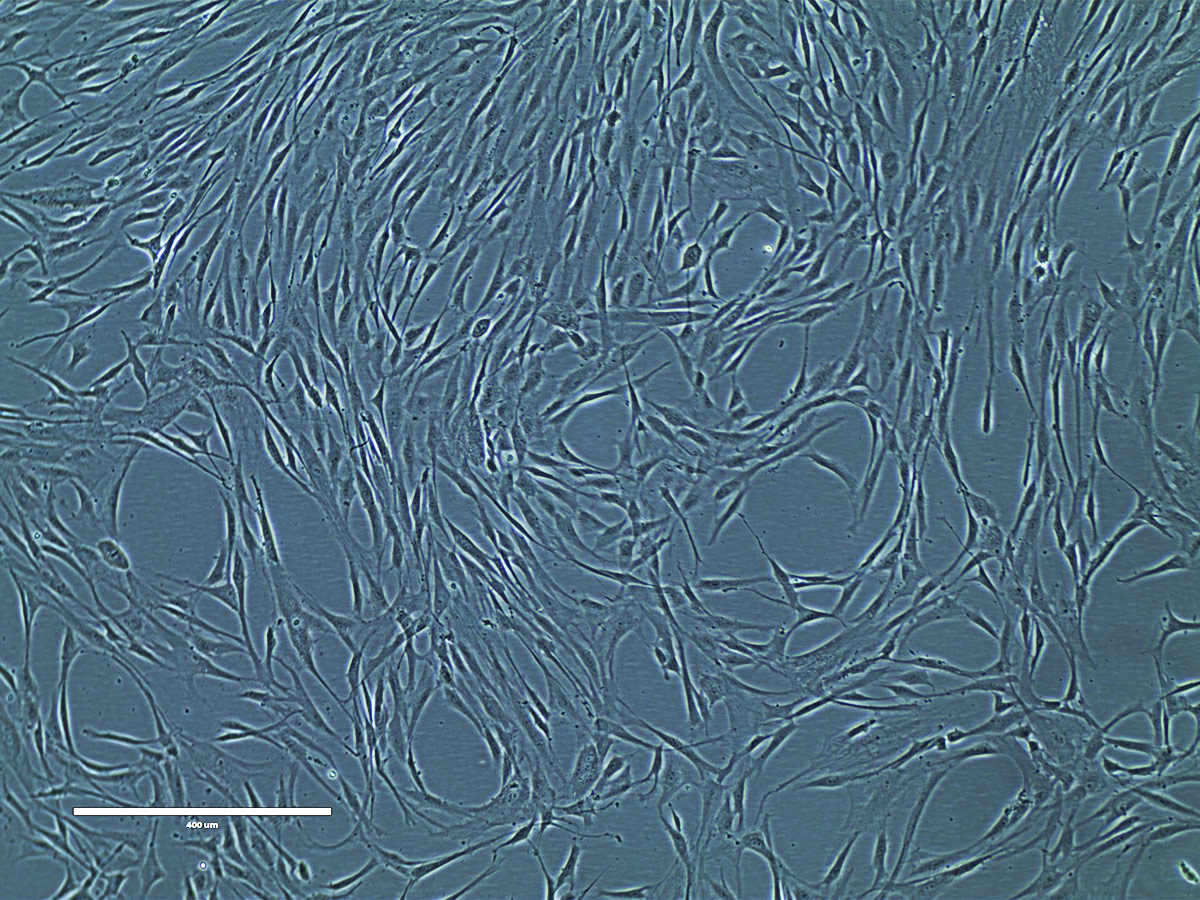
Figure 12. Phase contrast image of adipose tissue-derived MSCs at P2 (10x). Adipose tissue-derived MSCs (ATCC PCS-500-011) at passage 2 were seeded at 5,000 cells/cm2 in T-75 flasks and were cultured in Mesenchymal Stem Cell Basal Medium (ATCC PCS-500-030) supplemented with Mesenchymal Stem Cell Growth Kit - Low Serum (ATCC PCS-500-040) in a 37°C, 5% CO2 humidified incubator for 5 days. These cells are about 80% confluence ready for subculture.
Passaging
- Warm both the Trypsin-EDTA for Primary Cells (ATCC PCS-999-003) and the Trypsin Neutralizing Solution (ATCC PCS-999-004) to room temperature prior to dissociation. Warm the complete growth medium to 37°C prior to use with the cells.
- Aspirate the spent media carefully without disturbing the monolayer.
- Rinse the cell layer once with 3 to 5 mL D-PBS (ATCC 30-2200) to remove residual medium.
- Add pre-warmed trypsin-EDTA solution (1 to 2 mL for every 25 cm2) to each flask.
- Rock each flask gently to ensure complete coverage of the trypsin-EDTA solution over the cells.
- Observe the cells under the microscope. When the cells pull away from each other and round up (typically within 1 to 3 minutes), remove the flask from the microscope and gently tap it from several sides to promote detachment of the cells from the flask surface.
- When the majority of cells appear to have detached, quickly add an equal volume of the Trypsin Neutralizing Solution (ATCC PCS-999-004) to each flask. Gently pipette or swirl the culture to ensure all of the trypsin-EDTA solution has been neutralized.
- Transfer the dissociated cells to a sterile centrifuge tube and set aside while processing any cells that remain in the culture flask.
- Add 3 to 5 mL D-PBS (ATCC 30-2200) to the tissue culture flask to collect any additional cells that might have been left behind.
- Transfer the cell/D-PBS suspension to the centrifuge tube containing the trypsin-EDTA dissociated cells.
- Repeat steps 10 and 11 as needed until all cells have been collected from the flask.
- Centrifuge the cells at 150 x g for 3 to 5 minutes.
- Aspirate neutralized dissociation solution from the cell pellet and re-suspend the cells in 2 to 8 mL fresh, pre-warmed, complete growth medium.
- Count the cells and seed new culture flasks at a density of 5,000 viable cells per cm2.
- Place newly seeded flasks in a 37°C, 5% CO2 incubator for 24 to 48 hours before processing the cells further. Refer to the section on Maintenance for guidelines on feeding.
Cryopreservation of Mesenchymal Stem Cells
- Harvest MSCs as described above.
- Resuspend cells in complete MSC growth media into a single-cell suspension at cell concentration of 2 x 106 cells/mL.
- Add equal volume of 2x MSC freezing media (80% complete MSC growth media plus 20% DMSO, ATCC 4-X).
- Transfer 1 mL of the cell suspension (about 1 million cells/mL) into cryogenic storage vials (cryovials).
- Freeze the cells gradually at a rate of -1°C/min until the temperature reaches -70°C to -80°C. A cryopreservation container (eg, CoolCell freezing container, ATCC ACS-6000) may also be used.
- Transfer frozen cryovials to the vapor phase of liquid nitrogen for long-term storage.
Adipose-derived Mesenchymal Stem Cell Differentiation Protocols
Adipocyte Differentiation
The Adipocyte Differentiation Toolkit for Adipose-derived MSCs and Preadipocytes (ATCC PCS-500-50) contains medium and reagents designed to induce adipogenesis, with high efficiency, in actively proliferating Adipose-derived Mesenchymal Stem Cells (ATCC PCS-500-011), and support maturation of derived adipocytes during lipid accumulation.
ATCC also offers Chondrocyte (ATCC PCS-500-051) and Osteocyte (ATCC PCS-500-052) Differentiation Tools for Adipose-derived Mesenchymal Stem Cells, and the Adipocyte Differentiation Toolkit for Bone Marrow and Umbilical-derived MSCs (ATCC PCS-500-053). Please visit www.atcc.org/stemcells for additional information.
Preparing Cells for Adipocyte Differentiation
- Follow the instructions for the growth of Adipose-derived Mesenchymal Stem Cells (ATCC PCS-500-011). It is recommended that the cells not be passaged more than four times before initiating adipocyte differentiation.
- When cells are 70-80% confluent, passage them into a tissue culture plate at a density of 18,000 cells/cm2. Adjust the number of cells and volume of media according to the tissue culture plate used. Please see the example below.
Example: For a 6-well tissue culture plate with a surface area of 9.5 cm2/well, add a total of 171,000 viable cells to each well containing 2 mL of Mesenchymal Stem Cell Basal Medium (ATCC PCS-500-030) supplemented with Mesenchymal Stem Cell Growth Kit-Low Serum (ATCC PCS-500-040) components. - Rock the plate gently back and forth and side to side to distribute the cells evenly before incubation. Do not swirl.
- Incubate the cells in a 37°C, 5% CO2, incubator for 48 hours before initiating adipocyte differentiation.
Adipocyte Differentiation (AD) Media Preparation
The adipocyte differentiation process requires two separate media preparations: one for initiation of differentiation and one for maintenance. Stock solutions of these media can be prepared in tandem, in advance as follows:
- Thaw all three components of the differentiation kit and warm to 37°C in a water bath.
NOTE: It may be necessary to shake the AD Supplement and the ADM Supplement upon warming to help re-dissolve any components that may have precipitated out of solution upon freezing. - Decontaminate the external surfaces of all three kit components by spraying them with 70% ethanol.
- Using aseptic technique and working in a laminar flow hood or biosafety cabinet:
- Transfer 15 mL of Adipocyte Basal Medium and 1 mL of Adipocyte Differentiation (AD) Supplement to a sterile 50 mL conical tube, using a separate sterile pipette for each transfer. This is your working stock of Adipocyte Differentiation (AD) Initiation Medium used during the first 48 hours of differentiation.
- Add 5 mL of Adipocyte Differentiation Maintenance (ADM) Supplement to the remaining 85 mL of Adipocyte Basal Medium. This is your working stock of Adipocyte Differentiation Maintenance (ADM) Medium.
- Tightly cap each container of medium and swirl the contents gently to assure a homogeneous solution. Do not shake forcefully to avoid foaming. Label and date the bottle.
NOTE: It is recommended that you transfer the required volume of media to a sterile tube for prewarming prior to each feeding rather than repeatedly re-warming the entire working stock. - Each container of differentiation medium should be stored in the dark at 2°C to 8°C (do not freeze). When stored under these conditions, the differentiation medium is stable for up to three weeks.
Adipocyte Differentiation Procedure
A. Initiation Phase
- After incubating the prepared Adipose-derived Mesenchymal Stem Cells as described above, carefully aspirate the media from the wells.
- Immediately rinse the cells once by adding 2 mL of room-temperature D-PBS (ATCC 30-2200) to each well, then carefully aspirate the PBS from the wells.
- Add 2 mL of pre-warmed (37°C) Adipocyte Differentiation Initiation Medium to each well to begin the adipocyte differentiation process.
- Incubate the cells in a 37°C, 5% CO2 incubator for 48 hours.
- Feed the cells by carefully removing half the volume of medium (1 mL) from each well and adding another 2 mL of pre-warmed (37°C) Adipocyte Differentiation Initiation Medium to each well.
Important: DO NOT TILT the plate during aspiration. It is important that the cell monolayer is not exposed to air during this and subsequent steps to ensure that developing lipid vesicles do not burst.
B. Maintenance Phase
- Incubate the cells in a 37°C, 5% CO2 incubator for 48 hours.
- Carefully remove 2 mL of medium from each well (leaving 1 mL) and replace with 2 mL of pre- warmed (37°C) Adipocyte Differentiation Maintenance Medium.
Important: DO NOT TILT the plate during aspiration. It is important that the cell monolayer is not exposed to air during this and subsequent steps to ensure that developing lipid vesicles do not burst. - Repeat Steps 1 and 2 every 3-4 days for another 11 days until adipocytes reach full maturity. Full maturity will be reached 15 days after the beginning of initiation phase, or 17 days from initial plating of cells.
- Cells can be used at any phase of adipocyte differentiation as predicated upon experimental design. To confirm lipid accumulation, cells can be fixed and stained with Oil Red O.
ATCC Cancer Stem Cell Culture
Introduction
Cancer stem cells (CSCs) are a subpopulation of cancer cells seen in some cancer types that have the capacity to self-renew, proliferate, and differentiate into different cell types. These cells have also been shown to possess the ability to drive the formation and growth of the tumor. Given these properties, it is possible CSCs could be responsible for cancer metastases and relapse. Thus, cultured CSCs may be a powerful tool for discovering the next generation of oncology therapeutics.
ATCC recently acquired the CSC line BT142 mut/- (ATCC ACS-1018) from Drs. Samuel Weiss and Gregory Cairncross of the University of Calgary. This CSC line was originally isolated from a patient with grade III Oligoastrocytoma containing a heterozygous R132H mutation in the isocitrate dehydrogenase gene 1 (IDH1).19 Subsequent culturing of initially isolated cells resulted in the loss of wild-type allele of IDH1 while retaining the mutant R132H allele leading to the subsequent cell line BT142 mut/-. The loss of the wild-type allele in the BT142 mut/- cells resulted in the decrease of 2-hydroxyglutarate production similar to reports observed in vivo in patients.19 Since mutations in IDH1 are thought to be one of the earliest events in the development of glioma and R132H is the most common substitution, the BT142 mut/- cell line provides a model system for studying the biology of IDH1-mutant glioma and for the validation of compounds targeting IDH1-mutant cells.14
Protocol for Culturing
Materials and Preparation of Complete Growth Medium
There are two options of base media for culturing this cell line:
Option 1: NeuroCult NS-A Proliferation kit (catalog No. 5751, Stem Cell Technologies)
Option 2: Supplement DMEM/F12 (1:1) (ATCC 30-2006) with:
- 0.9% glucose
- 4 mM L-glutamine (ATCC 30-2214)
- 25 μg/mL insulin
- 100 μg/mL transferrin
- 20 nM progesterone
- 15 uM putrescine
- 30 nM selenite
To make the complete growth medium, add the following additional supplements to either option of the base medium:
- 20 ng/mL rh EGF
- 100 ng/mL recombinant human Platelet-Derived Growth Factor-AA (PDGF-AA, Catalog No. 100-13A, PeproTech)
- 20 ng/mL rh FGF
- 2 μg/mL heparan sulfate (Catalog No. H9902, Sigma)
Important: The ATCC ACS-1018 BT142 mut/- neurospheres may stick to plastic disposable tips and some plastic tubes. To prevent cell loss, we recommend that you pre-wet the pipette tip with media before drawing the cells up into the pipette. In addition, we recommend using polypropylene tubes when working with neurosphere cultures.
Thawing Procedure for BT142 mut/- cell line
Volumes used in this protocol are for a 75 cm2 flask.
- The cells are cryopreserved as neurospheres and should be thawed as clusters. Do not break apart the neurospheres into a single cell suspension.
NOTE: BT142 mut/- cells grow as phase- bright, smooth spheres. The neurospheres should not get too big, ragged or dark as this is a sign of unhealthy, dying cells. The cells should be passaged when the neurospheres are 200 to 400 μm in size. Figure 13. - Quickly thaw the vial by gentle agitation in a 37 °C water bath. To reduce the possibility of contamination, keep the O-ring and cap out of the water.
- Remove the vial from the water bath as soon as the contents are thawed, and decontaminate by spraying with 70% ethanol. All of the operations from this point on should be carried out under strict aseptic conditions.
- Transfer the contents of the vial to a centrifuge tube containing 10 mL of complete culture medium.
- Centrifuge the cells at 200 x g for 10 minutes.
- Aspirate supernatant and resuspend the cells in 15 mL of complete culture medium and dispense into a 75 cm2 flask.
- The cells may take a few weeks to recover from cryopreservation. Viable neurospheres are semi-transparent and phase contrast bright with smooth outer surfaces.
Maintenance of BT142 mut/- cultures
Volumes used in this protocol are for a 75 cm2 flask.
Replace medium once per week, or as required if media looks depleted (orange/yellow in color), by replacing 5 mL with fresh complete culture medium. It is important to monitor the culture regularly so the medium is changed when needed.
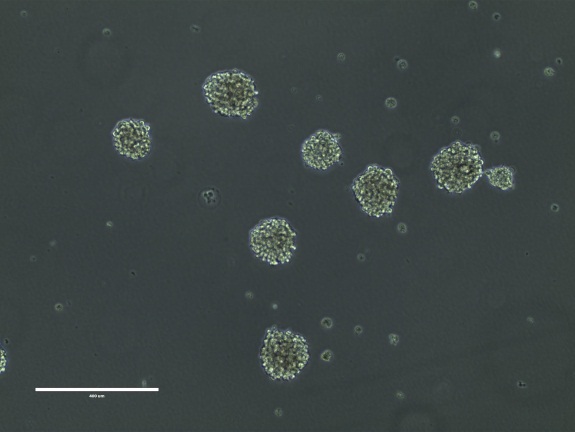
Figure 13. BT142 mut/- neurospheres. Bar = 400 μm
Passaging BT142 mut/- cultures
Volumes used in this protocol are for a 75 cm2 flask.
- Harvest and collect the entire cell suspension from the culture flask into a 15 mL tube.
- Centrifuge at 200 x g for 10 minutes.
- Aspirate supernatant, leaving approximately 200 μL to cover the pellet.
- Add 1 mL of complete culture medium.
- Triturate cells with a P1000 micropipette set to 800 μL by pipetting up and down 40 times or until the cells appear to be in a single cell suspension.
- Add 8 mL of complete culture medium and centrifuge at 200 x g for 10 minutes.
- Aspirate supernatant and resuspend the cells in 2 mL of complete culture medium.
- Count viable cells using trypan blue exclusion assay on a hemacytometer.
NOTE: If accurate cell count is necessary, Accumax (Catalog No. AM 105, Innovative Cell Technologies) can be used; however, the cells may take some time to recover from an enzymatic dissociation. - Seed single cells between ranges of 8 x 103 to 2 x 104 cells/cm2.
Cryopreservation of BT142 mut/- cell line
- Centrifuge neurospheres at 200 x g for 10 minutes.
- Resuspend in complete growth media by pipetting up and down 2 to 3 times. Take care not to disrupt neurospheres.
- Add equal volume of 2x freezing media (80% complete growth media plus 20% DMSO, ATCC 4-X) to the resuspended cells.
- Transfer the neurosphere suspension into cryovials.
- Freeze the cells gradually at a rate of -1°C/min until the temperature reaches -70°C to -80°C. A cryopreservation container (eg, CoolCell freezing container, ATCC ACS-6000) may also be used.
The cells should not be left at -80°C for more than 24 to 48 hours. Once at -80°C, frozen cryovials should be transferred to the vapor phase of liquid nitrogen for long-term storage.
Mouse Embryonic Stem Cell Culture Guide
Introduction
mESCs were first isolated and propagated in culture in 1981. mESCs are typically isolated from blastocysts from the inner cell mass of 3.5 day-old embryos and can be maintained in an undifferentiated, pluripotent state by culture with LIF on a feeder layer of mitotically-arrested mouse embryonic fibroblasts or in a feeder-free environment using gelatin-coated flasks.
Applications of mESCs
Being the first to be cultured for extended periods in an undifferentiated state while still maintaining their pluripotency, mESCs have provided the experimental foundation necessary for more recent stem cell innovations such as hESCs and iPSCs. More important, mESCs have proven to be invaluable in understanding many aspects of cell and developmental biology, including cell cycle regulation, cellular interactions during development, and the control of gene expression. mESCs will continue to be an important system for understanding stem cell and tissue biology and for creating transgenic or knock-out mice, used as in vivo disease models, by homologous recombination. Recent applications of mESCs include:
- Identifying factors that maintain pluripotency15
- Human genetic disease research16
- Identifying novel differentiation factors17
- Cancer biology18
ATCC Mouse Embryonic Stem Cell Solutions
ATCC offers 15 mESC lines, including lines that express Green or Yellow Fluorescent Protein (GFP/YFP) markers. In addition, ATCC provides a portfolio of cell culture media, sera, and reagents, such as Mouse ES Basal Medium (ATCC SCRR-2011), ES cell qualified FBS (ATCC SCRR-30-2020), and mouse embryonic fibroblast cell lines that may be used as feeder layers for the culture of mouse ES cells.
To ensure the purity of ATCC mESCs, each batch is isolated from a single mouse embryo of known genotype and tested for:
- Mycoplasma, bacterial, and yeast contamination.
- Authentication of growth and morphology, including adherence to plastic when cultured in optimized Mouse ES Basal Cell Media.
Protocols for Culturing mESCs
Materials
Table 7. Mouse ES Cells
| ATCC No. | Cell Line Designation | Applications/Markers Expressed |
|---|---|---|
| CRL-1934 | ES-D3 (D3) | Producing transgenic and knock-out mouse lines |
| SCRC-1002 | C57BL/6 | |
| SCRC-1010 | J1 | Gene expression studies, disease models |
| SCRC-1011 | R1 | |
| SCRC-1016 | ESF 158 | |
| SCRC-1018 | RW.4 | Producing transgenic and knock-out mouse lines |
| SCRC-1019 | B6/BLU | Contains a LacZ-β-globin reporter gene specifically expressed in red blood cells |
| SCRC-1020 | SCC#10 | Gene knock-out/knock-in |
| SCRC-1021 | EDJ#22 | Target gene mutation/knock out |
| SCRC-1023 | AB2.2 | Target gene mutation/knock out. Tissue engineering |
| SCRC-1033 | 7AC5/EYFP | Cells express EYFP for tracking stem cell differentiation during development |
| SCRC-1036 | R1/E | Cell differentiation studies |
| SCRC-1037 | G-Olig2 | Cells express GFP-Olig2 for lineage specific tracking |
| SCRC-1038 | CE-1 | Producing transgenic and knock-out mouse lines, cells are resistant to hygromycin |
| SCRC-1039 | CE-3 | Producing transgenic and knock-out mouse lines, neural-differentiated cell lineages express GFP marker, cells are resistant to hygromycin |
Table 8. Complete Growth Medium
| ATCC No./ Product No. | Product Name | Size | Storage |
|---|---|---|---|
| SCRR-2011 | Mouse ES Cell Basal Medium | 500 mL | 2°C to 8°C |
| SCRR-30-2020 | Fetal Bovine Serum, ES-Qualified | 500 mL | -70°C |
| 30-2101 | Trypsin-EDTA Solution | 100 mL | -20°C |
| 30-2300 | Penicillin - Streptomycin Solution | 100 mL | -20°C |
| EMD Millipore Cat. No. ESG1107 | Mouse leukemia inhibitory factor (LIF) | 1.0 mL | -20°C |
| Life Technologies Cat. No. 21985 | 2-mercaptoethanol | 50mL | 2°C to 8°C |
Preparation of Media Reagents
- Obtain one bottle of Mouse ES Cell Basal Medium from cold storage.
- Decontaminate the external surfaces of all growth kit component vials and the basal medium bottle by spraying them with 70% ethanol.
- Using aseptic technique and working in a laminar flow hood or biosafety cabinet, transfer the indicated volume of each component, as indicated in the following table, to the bottle of basal medium using a separate sterile pipette for each transfer.
| Component | Final Concentration |
|---|---|
| FBS, ES-Qualified (ATCC SCRR-30-2020) | 15% |
| LIF | 1,000 U/mL |
| 2-mercaptoethanol | 0.1 mM |
- Cap the bottle of complete growth medium and swirl the contents gently to assure a homogeneous solution. Do not shake forcefully to avoid foaming. Label and date the bottle.
- Complete growth media should be stored in the dark at 2°C to 8°C (do not freeze). When stored under these conditions complete growth media is stable for two weeks. This medium is formulated for use with a 5% CO2 humid incubator.
Complete Medium for Feeder Cells
Feeder cells (ATCC feeder cells are listed in the Appendix) may be grown in DMEM (ATCC 30-2002) and 10-15% FBS (ATCC 30-2020). Please note that feeder cell AFT024 (ATCC SCRC-1007) also requires that 2-mercaptoethanol be added to the growth medium to a final concentration of 0.05 mM. Please consult the product sheet provided for the feeder cell line you wish to use for specific medium requirements and subculturing procedures.
Feeder cells should be initiated 24-48 hours prior to inoculation with mESCs.
Thawing of Cryopreserved mESCs
- At least one day before plating the mESCs, prepare the desired combination of flasks with feeder cells to accommodate an initial ES cell seeding density of 30,000 cells/cm2 to 50,000 cells/cm2.
Refer to the batch specific information provided on the last page of the product information sheet for the total number of viable cells recovered from each lot of ATCC mESCs. - Plate mitotically arrested MEFs as a feeder layer at approximately 55,000 feeder cells/cm2 in complete medium for feeder cells. Refer to the product sheet for mitotically arrested MEF for detailed handling instructions.
Feeder cells should be used within one week of plating. It is best to use feeder cells within 24 or 48 hours of initiation.
Plating mESCs
- Pre-warm complete growth medium for mESCs at 37°C for at least 30 minutes before adding to cells.
- Perform a 100% medium change for the MEF feeder cells using Complete Growth Medium for mESCs one hour prior to thawing the mESCs.
- Thaw the vial of mESCs by gentle agitation in a 37°C water bath. To reduce the possibility of contamination, keep the O-ring and cap out of the water. Thawing should be rapid (approximately 90 seconds).
- Remove the vial from the water bath before the contents are completely thawed, and decontaminate by dipping in or spraying with 70% ethanol. All of the operations from this point on should be carried out under strict aseptic conditions.
- Transfer the contents of the vial plus 5 mL of complete growth medium for mESCs to a 15 mL centrifuge tube. Use an additional 1 mL of media to rinse the vial and transfer the liquid to the 15 mL tube. Add 4 mL of complete growth medium for mESCs to bring the total volume to 10 mL.
- Spin the cells at 270 x g for 5 min. Aspirate the supernatant and resuspend the pellet in 2 mL of complete growth medium for mESCs.
- Add the 2 mL of cell suspension to the appropriate size flask containing feeder cells and fresh complete growth medium for mESCs (see batch specific information). mESCs should be plated at a density of 30,000 - 50,000 cells/ cm2.
- Incubate the culture at 37°C, 5% CO2, humid incubator.
Routine Handling of mESC Cultures
Perform a 100% medium change every day. Passage the cells every 1 to 2 days. If the colonies are close to, or touching each other the culture is overgrown. Overgrowth will result in differentiation.
Make sure that you have prepared a sufficient number of flasks pre-plated with MEF feeder layers to support frequent passage of the mESCs.
Passaging
Feeder Cell Preparation for Subcultures
- Maintain a sufficient number of flasks each day that have been pre-plated with MEFs in complete medium for feeder cells.
- One hour before subculturing the mESCs, perform a 100% medium change for the MEFs using complete growth medium for mESCs.
NOTE: To ensure the highest level of viability, pre-warm media and Trypsin/EDTA to 37°C before adding to cells. Volumes used in this protocol are for T75 flasks. Proportionally adjust the volumes for culture vessels of other sizes. A split ratio of 1:4 to 1:7 is recommended.
Dissociation and Transfer of mESCs
- Aspirate the medium from the flask(s) containing mESCs.
- Wash with PBS Ca+2/Mg+2-free (ATCC SCRR-2201).
- Add 3.0 mL of 0.25% (w/v) Trypsin / 0.53 mM EDTA solution (ATCC 30-2101) and place in incubator. After about one minute the mESC colonies will dissociate and all cells will detach from the flask.
- Dislodge the cells by gently tapping the side of the flask then wash the cells of with 7 to 10 mL of fresh culture medium. Triturate cells several times with a 10 mL pipette in order to dissociate the cells into a single-cell suspension. Spin the cells at 270 x g for 5 min. Aspirate the supernatant.
- Resuspend in enough complete growth medium for mESCs to reseed new vessels at the desired split ratio (a split ratio of 1:4 to 1:7 is recommended). Perform a cell count to determine the total number of cells. ES cells should be plated at a density of 30,000 – 50,000 cells/ cm2.
- Add separate aliquots of the cell suspension to the appropriate size flask containing feeder cells and add an appropriate volume of fresh complete growth medium for mESCs to each vessel.
- Incubate the culture at 37°C, 5% CO2, humid incubator. Perform a 100% medium change every day, passage cells every 1 to 2 days.
Cryopreservation of mESCs
- Harvest mouse mESCs including mouse feeder cells described above.
- Resuspend cells in complete growth medium for mESCs into a single-cell suspension at cell concentration of 2 x 106 cells/mL.
- Add equal volume of 2x mESCs freezing media (80% complete mESCs growth media plus 20% DMSO, ATCC 4-X).
- Transfer 1 mL of the cell suspension (about 1 x 106 cells/mL) into cryovials.
- Place cryovials into freezing containers and store overnight at a -70°C to -80°C freezer.
- Transfer frozen cryovials to the vapor phase of liquid nitrogen for long-term storage.
For additional information about ATCC mESCs, please visit www.atcc.org/stemcells.
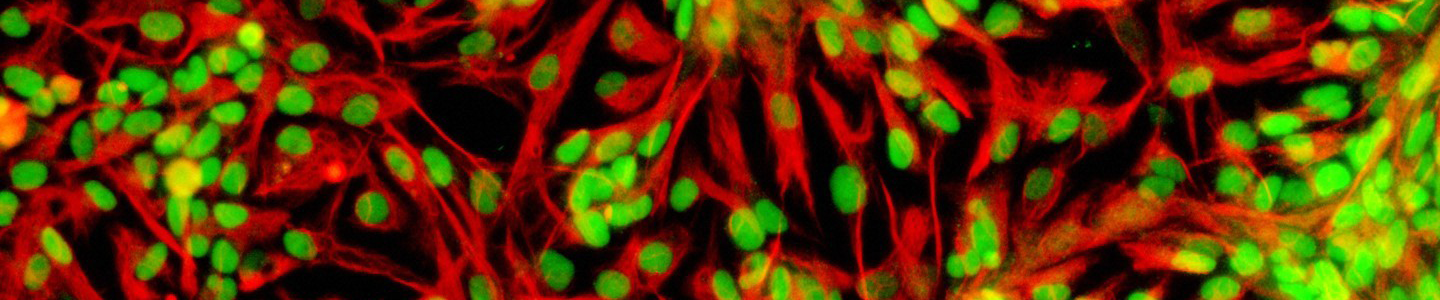
Appendix
Table 9. Recommended Volume of Media and Reagents for Different Cell Culture Dishes
| Cell Culture Vessel | Growth Area (cm2) | Pluripotent Stem Cell Culture Medium | Cell Basement Membrane | Feeder Medium (DMEM + 15% FBS) | Stem Cell Dissociation Reagent working solution | Stem Cell Freezing Media |
|---|---|---|---|---|---|---|
| 6 well /35 mm2 | 9.5 | 2 mL | 1 mL | 2 mL | 1 mL | 1 mL |
| 6 cm2 | 21 | 4 mL | 2 mL | 4 mL | 2 mL | 2 mL |
| 10 cm2 | 56 | 10 mL | 4 mL | 10 mL | 4 mL | 4 mL |
Table 10. ATCC iPSC lines
| ATCC No. | Designation | Reprogramming Method | Tissue of Origin | Disease | Ethnicity | Gender |
|---|---|---|---|---|---|---|
| ACS-1003 | ATCC-DYP0730 | Episomal | Foreskin | Down syndrome | Caucasian | Male |
| ACS-1004 | ATCC-DYP0250 | Episomal | Dermal fibroblast | Cystic fibrosis; homozygous for the Delta 508 mutation in the CFTR gene | Unknown | Male |
| ACS-1007 | ATCC-HYR0103 | Retroviral | Liver | Normal | Hispanic | Male |
| ACS-1011 | ATCC-DYR0100 | Retroviral | Foreskin | Normal | Unknown | Male |
| ACS-1012 | ATCC-DYR0530 | Retroviral | Skin dermal fibroblast | Parkinson’s disease, asthma, depression | Caucasian | Male |
| ACS-1013 | ATCC-DYS0530 | Sendai viral | Skin dermal fibroblast | Parkinson’s disease, asthma, depression | Caucasian | Male |
| ACS-1014 | ATCC-DYP0530 | Episomal | Skin dermal fibroblast | Parkinson’s disease, asthma, depression | Caucasian | Male |
| ACS-1019 | ATCC-DYS0100 | Sendai viral | Foreskin | Normal | Unknown | Male |
| ACS-1020 | ATCC-HYS0103 | Sendai viral | Liver | Normal | Hispanic | Male |
| ACS-1021 | ATCC-CYS0105 | Sendai viral | Heart | Normal | Unknown | Male |
| ACS-1023 | KYOU- DXR0109B | Retroviral | Dermal fibroblast | Normal | Caucasian | Female |
| ACS-1024 | ATCC-BYS0110 | Sendai viral | Bone marrow CD34+ | Normal | African American | Male |
| ACS-1025 | ATCC-BYS0111 | Sendai viral | Bone marrow CD34+ | Normal | Hispanic | Male |
| ACS-1026 | ATCC-BYS0112 | Sendai viral | Bone marrow CD34+ | Normal | Non-Hispanic White | Male |
| ACS-1027 | ATCC-BYS0113 | Sendai viral | Bone marrow CD34+ | Normal | Asian | Male |
| ACS-1028 | ATCC-BXS0114 | Sendai viral | Bone marrow CD34+ | Normal | African American | Female |
| ACS-1029 | ATCC-BXS0115 | Sendai viral | Bone marrow CD34+ | Normal | Hispanic | Female |
| ACS-1030 | ATCC-BXS0116 | Sendai viral | Bone marrow CD34+ | Normal | Non-Hispanic White | Female |
| ACS-1031 | ATCC-BXS0117 | Sendai viral | Bone marrow CD34+ | Normal | Asian | Female |
Feeder Cell Lines
Table 11. Feeder Cell Lines: Qualified for Stem Cell Research (Inactivated)
| ATCC No. | Cell Line Designation | Description |
|---|---|---|
| SCRC-1007.1 | AFT024 IRR | Irradiated mouse embryonic liver fibroblast |
| SCRC-1008.1 | MEF (C57BL/6) IRR | Irradiated mouse embryonic fibroblast |
| SCRC-1040.1 | MEF (CF-1) IRR | Irradiated mouse embryonic fibroblast |
| SCRC-1040.2a | MEF (CF-1) MITC | Mitomycin C-treated mouse embryonic fibroblast |
| SCRC-1041.1 | HFF-1 IRR | Irradiated human foreskin fibroblast |
| 56-X.2 | MITC-STO | Mitomycin C –treated mouse embryonic fibroblast |
Table 12. Feeder Cell Lines: Qualified for Stem Cell Research (Not Inactivated)
| ATCC No. | Cell Line Designation | Description |
|---|---|---|
| SCRC-1007 | AFT024 | Mouse embryonic liver fibroblasts |
| SCRC-1008 | MEF (C57BL/6) | Mouse embryonic fibroblasts |
| SCRC-1040 | MEF (CF-1) | Mouse embryonic fibroblasts1 |
| SCRC-1041 | HFF-1 | Human foreskin fibroblasts |
| SCRC-1045 | MEF (DR4) | Multidrug resistant mouse fibroblasts |
| SCRC-1049 | SNL76/7 | STO fibroblasts with G418 resistance and endogenous expression of LIF2 |
| SCRC-1050 | SNLP 76/7-4 | STO fibroblasts with resistance to G418 and puromycin plus endogenous expression of LIF3 |
1If the MEFs are being used as a feeder layer for mESCs, it is not recommended to use them past passage no.6.
2Resistant to G418 (neomycin): 350 μg/mL. Cells express high levels of LIF.
3SNLP 76/7-4 is a puromycin-resistant derivative of SNL76/7.
4Mitomycin C is an antineoplastic antibiotic that inhibits DNA synthesis by cross-linking DNA at 5΄–CpG–3΄ sequences. It produces oxygen radicals and is preferentially toxic to hypoxic cells. Care must be taken when preparing the Mitomycin C solution. The fibroblasts must be extensively washed following treatment to prevent toxic effects to embryonic stem (ES) cells, or other cell types that require the use of a feeder cell layer for propagation.
Disclaimer: Products in this stem cell culture guide are not provided with a license to any patents not licensed to or owned by ATCC. User acknowledges and agrees that use of certain products in this culture guide may require a license from a person or entity other than ATCC, or be subject to restrictions that may be imposed by a person or entity other than ATCC.
Get the ATCC guide to culturing stem cells
Download the Guide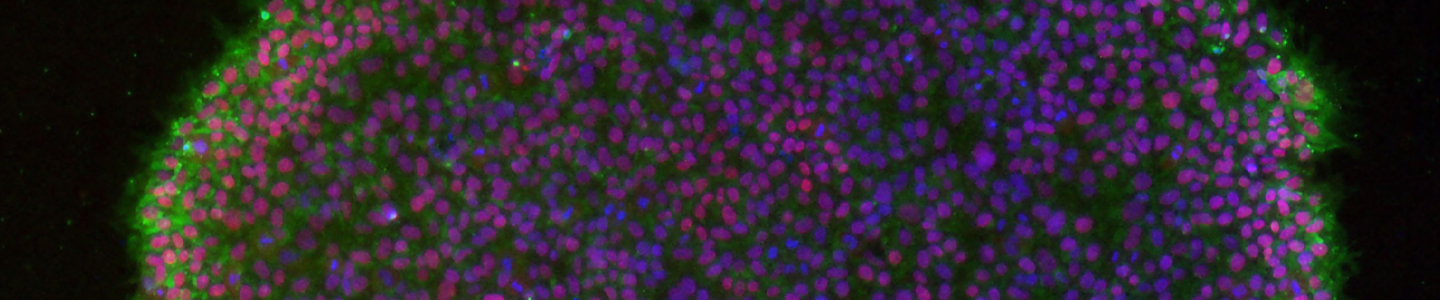
References
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126(4): 663-676, 2006. PubMed: 16904174
- Okita K, Yamanaka S. Induced pluripotent stem cells: opportunities and challenges. Philos Trans R Soc Lond B Biol Sci 366(1575):2198-207, 2011. PubMed: 21727125
- Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861-72 2007.
- Friedenstein AJ, et al. Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation 6: 230 –247, 1968. PubMed: 5654088
- Dominici M, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8(4): 315-317, 2006. PubMed: 16923606
- Zuk PA, et al. Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell 13(12): 4279- 4295, 2002. PubMed: 12475952
- Freshney RI. Culture of Animal Cells: A Manual of Basic Technique, 6th Ed. John Wiley & Sons, Hoboken, New Jersey, 2010.
- Schäffler A and Büchler C. Concise review: adipose tissue-derived stromal cells--basic and clinical implications for novel cell-based therapies. Stem Cells 25(4):818-827, 2007. PubMed: 17420225
- Seo MJ, et al. Differentiation of human adipose stromal cells into hepatic lineage in vitro and in vivo. Biochem Biophys Res Commun 328(1):258-264, 2005. PubMed: 15670778
- Kang S, et al. Neurogenesis of Rhesus adipose stromal cells. J Cell Sci 118: 4289-4299, 2004. PubMed: 15292397
- Safford KM, Rice HE. Stem cell therapy for neurologic disorders: therapeutic potential of adipose- derived stem cells. Curr Drug Targets 6(1): 57-62, 2005. PubMed: 15720213
- Kingham PJ, et al. Adipose-derived stem cells differentiate into a Schwann cell phenotype and promote neurite outgrowth in vitro. Exp Neurol. 207(2): 267-74, 2007 PubMed: 17761164
- Xu Y, et al. Neurospheres from rat adipose-derived stem cells could be induced into functional Schwann cell-like cells in vitro. BMC Neurosci Feb 12, 2008. PubMed: 18269732
- Luchman HA, et al. An in vivo patient-derived model of endogenous IDH1-mutant glioma. Neuro Oncol 14:184-191, 2012. PubMed: 22166263
- Abranches E, et al. Generation and characterization of a novel mouse embryonic stem cell line with a dynamic reporter of nanog expression. PloS One 8(3): e59928, Mar 19, 2013. PubMed: 23527287
- Chen J, et al. Mcph1-Defcient Mice Reveal a Role for MCPH1 in Otitis Media. PlosOne 8(3) e58156, Mar 13, 2013. PubMed: 23516444
- Park S, et al. Direct Effect of Chenodeoxycholic Acid on Differentiation of Mouse Embryonic Stem Cells Cultured under Feeder-free Culture Conditions. Biomed Res Int, Dec 29, 2012. PubMed: 23509715
- Li C, et al. Arf tumor suppressor and miR-205 regulate cell adhesion and formation of extraembryonic endoderm from pluripotent stem cells. Proc Natl Acad Sci USA 110(12):E112-21, 2013. PubMed: 23487795
- Luchman HA, et al. An in vivo patient-derived model of endogenous IDH1-mutant glioma. Neuro Oncol 14:184-91, 2012.